Журнал "Медицинский совет" №10/2022
DOI: 10.21518/2079-701X-2022-16-10-84-95
Т.Ю. Демидова1*, https://orcid.org/0000-0001-6385-540X
К.Г. Лобанова1, https://orcid.org/0000-0002-3656-0312
Н.С. Шевцова1, https://orcid.org/0000-0002-8344-6424
Т.Н. Короткова2, https://orcid.org/0000-0002-3684-9992
А.С. Кочина1, https://orcid.org/0000-0002-6826-5924
1 Российский национальный исследовательский медицинский университет имени Н.И. Пирогова; 117997, Россия, Москва, ул. Островитянова, д. 1
2 Федеральный исследовательский центр питания, биотехнологии и безопасности пищи; 115446, Россия, Москва, Каширское шоссе, д. 21
Инсулинорезистентность (ИР) – важная проблема человечества, ведущая к развитию многих метаболических нарушений. Патогенетический механизм развития ИР в настоящее время полностью не изучен. Тем не менее существует ряд гипотез, объясняющих развитие данного состояния. К ним относятся такие гипотезы, как гипотеза бережливого генотипа, бережливого фенотипа, гормональная, стрессовая, хороших и плохих калорий, хронического метаболического воспаления, микробиотическая и комплексная модель, предложенная профессором Райнером Штраубом. В данной статье подробно рассмотрена микробиотическая теория, которая объясняет механизм развития нечувствительности периферических тканей к инсулину при дисбиозе за счет увеличения трансмиссии провоспалительных молекул из кишечника в кровоток и активации системного воспаления, нарушения механизма «кишечник – мозг – периферия» и нарушения рецепторных взаимодействий активных метаболитов кишечной микробиоты (КМ) на уровне клеток метаболических органов. Ценность данной теории состоит в том, что ее факторы воздействуют на все звенья патогенеза развития ИР, отраженные в интегрированной комплексной модели профессора Штрауба. В обзоре подробно рассмотрено взаимовлияние КМ и метаболических процессов организма человека на развитие ИР, приведены данные клинических исследований влияния КМ (ее состава, активных метаболитов, отдельных штаммов бактерий) на развитие ИР и роли хронического метаболического воспаления в данном процессе. Кроме этого, уделено внимание двунаправленным влияниям КМ и метформина, приведены данные клинических исследований об изменении КМ здоровых людей и людей с ИР под воздействием метформина. Рассмотрено, как КМ влияет на фармакокинетику данного препарата. Также показана возможность коррекции ИР путем использования пищевых волокон.
Для цитирования: Демидова Т.Ю., Лобанова К.Г., Шевцова Н.С., Короткова Т.Н., Кочина А.С. Влияние кишечной микробиоты на развитие инсулинорезистентности. Медицинский Совет. 2022;(10):84-95. https://doi.org/10.21518/2079-701X-2022-16-10-84-95
Конфликт интересов: авторы заявляют об отсутствии конфликта интересов.
Influence of gut microbiota on the development of insulin resistance
Tatiana Yu. Demidova1*, https://orcid.org/0000-0001-6385-540X
Kristina G. Lobanova1, https://orcid.org/0000-0002-3656-0312
Nadezhda S. Shevtsova1, https://orcid.org/0000-0002-8344-6424
Tatiana N. Korotkova2, https://orcid.org/0000-0002-3684-9992
Anna S. Kochina1, https://orcid.org/0000-0002-6826-5924
1 Pirogov Russian National Research Medical University; 1, Ostrovityanov St., Moscow, 117997, Russia
2 Federal Research Center of Nutrition, Biotechnology and Food Safety; 21, Kashirskoye Shosse, Moscow, 115446, Russia
Insulin resistance (IR) is an important problem of humanity, which leads to development of many metabolic disorders. Сurrently the pathogenic mechanism of the development of IR is not completely investigated. Nevertheless, there are some hypotheses explaining the development of this condition. These include such hypotheses as the hypothesis of thrifty genotype, thrifty phenotype, hormonal, stress, good and bad calories, chronic metabolic inflammation, microbiotic and integrated model suggested by Professor Rainer Straub. In this article, the microbiotic theory will be considered in detail, explaining the mechanism of the development of peripheral tissue insensitivity to insulin in dysbiosis due to amplification of transmission by proinflammatory molecules from the intestine to the bloodstream and activation of systemic inflammation, disruption of the “gut-brain-periphery” mechanism and impaired receptor interactions of active intestinal metabolites of the gut microbiota (GM) at the level of cells of metabolic organs. The value of this theory is that its factors affect all links in the pathogenesis of the development of IR, reflected in the integrated model of Professor Straub. In this review the influence of GM and metabolic processes of human body on the development of IR will be considered in detail, data from clinical studies about the influence of GM (its composition, active metabolites, individual bacterial strains) on the development of IR and the role of chronic metabolic inflammation in this process will also be presented. In addition, attention will be paid to bidirectional effects of GM and metformin, as well as to data from clinical studies on changes in GM in healthy people and people with IR under the influence of metformin and how GM affects the pharmacokinetics of this drug. The possibility of IR correction through the use of dietary fiber will also be considered.
For citation: Demidova T.Yu., Lobanova K.G., Shevtsova N.S., Korotkova T.N., Kochina A.S. Influence of gut microbiota on the development of insulin resistance. Meditsinskiy sovet = Medical Council. 2022;(10):84-95. https://doi.org/10.21518/2079-701X-2022-16-10-84-95
Conflict of interest: the authors declare no conflict of interest.
Введение
Впервые термин «инсулинорезистентность» (ИР) был введен H.P. Himsworth и R.B. Kerr для обозначения недостаточного ответа на экзогенное введение инсулина у пациентов с сахарным диабетом 2-го типа (СД2) и ожирением. В дальнейшем он стал применяться для определения состояния, сопровождающегося устойчивостью тканей к сахароснижающему действию инсулина. Однако в настоящее время в связи с пониманием того, что инсулин регулирует метаболизм не только углеводов, но еще и белков и жиров, а также контролирует функции эндотелия и экспрессию ряда генов, под термином ИР принято понимать комбинированное изменение параметров метаболизма и митогенных процессов [1]. Так, согласно Американской диабетической ассоциации, ИР – это нарушение комплексного биологического ответа (метаболического и молекулярно-генетического) на инсулин (экзогенный и эндогенный), нарушение метаболизма углеводов, жиров, белков, а также изменения в синтезе дезоксирибонуклеиновой кислоты, регуляции транскрипции генов, процессов дифференцировки и роста клеток и тканей организма [2]. При развитии ИР на уровне основных метаболических тканей (мышечной, жировой и печеночной) нарушается транспорт глюкозы в клетки, активируются липолиз, гликогенолиз, глюконеогенез и катаболизм белков. Это приводит к повышению уровня глюкозы в крови, накоплению в крови триацилглицеридов, депонирующихся в печени и мышцах. Так, в ситуации недостатка основного энергетического ресурса клетки начинают использовать альтернативные источники энергии для своей жизнедеятельности [3].
В связи с этими метаболическими нарушениями происходят сбои в работе многих специализированных путей, что играет ведущую роль в развитии полиорганной дисфункции. Так, в состоянии ИР нарушается релаксация гладкомышечных клеток артерий из-за дефицита энергии, необходимой для активации путей синтеза оксида азота (NO). Это ведет к спазму сосудистой стенки и, как следствие, к ее повреждению. Поэтому ИР часто сопровождают артериальная гипертензия (АГ) и атеросклеротический процесс. В почках данную ситуацию усугубляет гиперактивация ренин-ангиотензин-альдостероновой системы, стимулирующая развитие вазоконстрикции выносящей артериолы, что приводит к развитию клубочковой гипертензии [4]. На уровне адипоцитов в условиях дефицита энергии активируется липолиз, который приводит к повышению количества свободных жирных кислот (СЖК) в крови. СЖК накапливаются в печени, развиваются стеатогепатит и неалкогольная жировая болезнь печени [5]. В кишечнике в условиях ИР развивается дисбиоз, снижаются синтез короткоцепочечных жирных кислот (КЦЖК) и продукция глюкагоноподобного пептида-1 (ГПП-1) L-клетками [6].
Однако более важным следствием ИР является развитие углеводных нарушений. Гипергликемия возникает в результате того, что на фоне ИР нарушается активация каскада внутриклеточных белков, ведущая к резкому снижению транспортной способности белков-переносчиков 4-го типа (GLUT-4), расположенных в первую очередь в мембране адипоцитов и миоцитов. В результате глюкоза не устремляется внутрь клеток, и развивается внутриклеточный дефицит энергии [4]. Для преодоления нечувствительности периферических тканей к действию инсулина β-клетки поджелудочной железы (ПЖЖ) увеличивают продукцию инсулина. Это приводит к избыточному накоплению энергии в инсулин-зависимых тканях. С течением времени ввиду длительной гипергликемии развивается компенсаторная ИР, из-за чего в крови повышается уровень и глюкозы, и инсулина. Таким образом, формируется порочный круг: высокий уровень инсулина ведет к ИР тканей, а ИР ведет к повышению глюкозы крови, в ответ на который повышается инсулин. К тому же избыточное накопление энергии в зависимых тканях увеличивает количество висцерального жира, что способствует его гиперваскуляризации и повышенному синтезу провоспалительных цитокинов в жировой ткани на фоне снижения продукции адипонектина, отвечающего за чувствительность тканей к инсулину [4, 6]. В конечном счете хроническое течение гипергликемии приводит к стрессу эндоплазматического ретикулума и истощению β-клеток ПЖЖ. Продукция инсулина прогрессивно снижается, развивается СД2 [5].
Теории развития инсулинорезистентности
Механизмы развития нечувствительности периферических тканей к инсулину до конца не изучены. Поэтому чтобы понять, какие основные факторы ведут к развитию ИР, рассмотрим наиболее известные теории ее возникновения и исследования, подтверждающие или опровергающие их.
Первая теория. Гипотеза бережливого генотипа
Первой теорией, объясняющей механизм развития ИР на уровне жировой и мышечной ткани, является гипотеза бережливого генотипа, описанная в 1962 г. Джеймсом Нилом. Согласно ей, у населения, генетически адаптированного к среде с дефицитом калорий, в геноме имеется ген бережливости, который способствует накоплению энергии в период изобилия, чтобы в дальнейшем ее использовать в период голодания. Этот ген включает быструю активацию гликогенеза и липогенеза путем повышенного синтеза инсулина в момент потребления пищи. Благодаря этому происходит накопление достаточных для длительных голоданий запасов энергетических ресурсов. В период голодания благодаря липолизу в крови накапливаются жирные кислоты и кетоновые тела, что ведет к снижению утилизации глюкозы тканями и, как следствие, к ИР. Эта ИР способствует меньшей утилизации оставшейся в крови глюкозы, которая в период голодания необходима для обеспечения энергией нервной и иммунной систем [7].
Существуют данные, подтверждающие и опровергающие данную гипотезу. С одной стороны, в 1999 г. группой ученых во главе с Хегеле было проведено исследование, в ходе которого была выявлена некоторая генетическая мутация, ассоциированная с гипергликемией среди коренных народов Сэнди-Лэйк, что позволило объяснить высокую распространенность СД2 у жителей провинции Онтарио в Канаде. Авторами исследования было сделано предположение, что данная мутация была следствием наличия у данной народности гена бережливости, который в прошлом помогал жителям провинции выживать в период длительного голодания [8]. Однако позже сам Хегеле усомнился в своих выводах. Он пришел к заключению, что мутация одного гена не может привести к развитию СД2 даже в период изобилия и в эру малоподвижного образа жизни [9]. Позже многие генетические исследования по всему миру опровергли наличие гена бережливости. Так, по данным исследования Q. Ayub et al., в котором проводилась оценка 65 локусов образцов африканского, европейского и восточноазиатского происхождения, не было обнаружено признаков положительного отбора гена бережливости [10]. К тому же J.R. Speakman et al. построили математическую модель, опровергающую существование гена бережливости. Согласно этой модели, если бы данный ген действительно существовал, он непременно достиг бы эволюционной фиксации: данный аллель считался бы наиболее выгодным, потому что увеличивал продолжительность жизни в период голодания из-за запасания энергии [11]. Таким образом, данная теория в XXI в. утратила свою актуальность в связи с отсутствием научных подтверждений.
Вторая теория. Гипотеза бережливого фенотипа
Следующей теорией развития ИР является теория бережливого фенотипа. Согласно ей, при материнском голодании в период беременности возникает дефицит питания плода, ведущий к гемодинамическим, метаболическим, гормональным изменениям. В дальнейшем эти изменения могут способствовать развитию у новорожденного эндокринных нарушений, приводящих во взрослой жизни к ряду заболеваний, таких как СД2, хроническая болезнь почек, идиопатическая АГ, ожирение и др. Дело в том, что одним из последствий дефицита питания является ИР, которая объясняется физиологической адаптацией плода к дефициту глюкозы и которая сохраняется на всю жизнь [12].
Правомочность данной теории подтверждает исследование C.N. Hales et al., в котором у потомства самок крыс, которых в период беременности кормили низкокалорийной низкобелковой пищей, демонстрируется снижение чувствительности гепатоцитов к действию инсулина и глюкагона [13]. Однако данная теория не учитывает других механизмов возникновения ИР у плода, которые могут оказаться более значимыми. Так, в исследовании Y. Nakano демонстрируется снижение количества уровней общего и высокомолекулярного адипонектина в сыворотке у недоношенных детей по сравнению с доношенными [14], а ведь адипонектин секретируется адипоцитами и играет ключевую роль в чувствительности тканей к инсулину. При этом высокомолекулярный адипонектин является его активной формой. Количество адипоцитов у недоношенных детей снижено, поэтому в дальнейшем формируется дисфункция жировой ткани и развивается ИР [14]. Так, гипотеза бережливого фенотипа объясняет только один путь формирования ИР у плода с дефицитом питания, поэтому мы не можем считать ее до конца правомерной.
Третья теория. Гормональная гипотеза
Следующей является гормональная теория ИР, которая гласит, что дисбаланс между контринсулярными гормонами и инсулином, возникающий при определенных эндокринных патологиях, приводит к таким метаболическим нарушениям, как увеличение аппетита, торможение липогенеза, стимуляция глюконеогенеза, снижение чувствительности тканей к инсулину и др. К таким патологиям относятся синдром/болезнь Иценко – Кушинга (повышение кортизола), акромегалия (повышение гормона роста), феохромацитома (повышение катехоламинов), глюкагонома (повышение глюкагона) и др. [2]. Так, группа ученых во главе с V. Guarnotta среди 192 пациентов с активным эндогенным синдромом Кушинга провели поперечное исследование, которое выявило нарушение метаболизма глюкозы у всех исследуемых, включая тех, чей уровень кортизола был повышен незначительно [15]. Однако стоит учитывать, что эта гипотеза демонстрирует лишь патологический путь формирования ИР. При рациональной терапии гормональный дисбаланс поддается коррекции с последующим восстановлением должного уровня инсулина и снижением ИР тканей-мишеней.
Четвертая теория. «Стрессовая» гипотеза
Еще одной причиной развития ИР является активация стресс-систем. Не секрет, что хронический стресс, психические заболевания, голодание, острое воспаление способствуют развитию ИР. Это происходит в связи с активацией оси «гипоталамус – гипофиз – надпочечники» в ответ на раздражители, что ведет к секреции адренокортикотропного гормона (АКТГ), стимулирующего синтез кортизола надпочечниками [16]. Из-за повышенного содержания кортизола в крови синтез кортиколиберина, стимулирующего выход инсулина из β-клеток ПЖЖ, снижается. К тому же кортизол вызывает повышенный синтез провоспалительных цитокинов, тормозящих экспрессию GLUT-4 на мембранах адипоцитов и миоцитов [17]. Совокупность этих факторов приводит к ИР. Прямая связь между уровнем кортизола и СД2 продемонстрирована в исследовании A. Steptoe et al., где количество кортизола в слюне у диабетиков было на 36% выше по сравнению с контрольной группой. Более того, пациенты с СД2 чаще сообщали о наличии депрессивных симптомов по сравнению со здоровыми [18]. Однако опять же существует множество других путей развития ИР, не зависящих от факторов стресса, поэтому эта гипотеза лишь частично раскрывает причины ИР.
Пятая теория. «Плохие и хорошие» калории
Также существует теория хороших и плохих калорий, объясняющая влияние различных диет на развитие ИР. Согласно данной теории, диета, богатая ненасыщенными жирами, улучшает чувствительность тканей к инсулину, снижает аппетит. Высокоуглеводная диета, напротив, ведет к избыточному постпрандиальному повышению инсулина и, как следствие, к ИР [19]. Секрет полиненасыщенных жирных кислот в том, что они обладают противовоспалительным потенциалом и к тому же повышают синтез ГПП-1. Благодаря этому уровень постпрандиальной гликемии быстро корректируется, и чрезмерного синтеза инсулина не возникает [20]. Подтверждает данную гипотезу исследование M.D. Gadgil et al., в котором диета, богатая ненасыщенными жирами по сравнению с высокоуглеводной и высокобелковой диетами, была ассоциирована с более низким уровнем ИР, оцениваемым по индексу количественной проверки чувствительности к инсулину (QUICKI) [19].
Шестая теория. Гипотеза хронического метаболического воспаления
Еще одной теорией ИР является теория хронического метаболического воспаления. Стоит уточнить, что в данном контексте метаболическое воспаление – это состояние, возникающее при нарушении глюкометаболических путей у пациентов с ожирением и СД2, сопровождающееся увеличением воспалительных цитокинов [21]. Эти провоспалительные цитокины снижают экспрессию GLUT-4 и способствуют развитию ИР [3]. Так, в исследовании A.E. Iglesias Molli et al. наблюдается прямая зависимость между наличием метаболического синдрома и ИР. Ученые сравнивают показатели индекса ИР (HOMA-IR) и С-реактивного белка (СРБ) у трех групп: метаболически здоровых без ожирения (первая группа), метаболически здоровых с ожирением (вторая группа) и пациентов с ожирением и метаболическим синдромом (третья группа). Уровень СРБ примерно одинаково повышен у пациентов второй и третьей групп, тогда как среди представителей первой группы он в норме. Несмотря на это, HOMA-IR, подтверждающий наличие ИР, оказался повышен только в группе пациентов, имеющих как ожирение, так и другие компоненты метаболического синдрома [22]. Данная гипотеза, как и предыдущие, рассматривает только один из путей формирования ИР, поэтому мы не можем считать ее единственно верной и полноценной.
Комплексная теория. Интегрированная модель Райнера Штрауба
В настоящее время наиболее актуальной является комплексная интегрированная модель ИР, предложенная профессором Райнером Штраубом (Германия). Суть этой теории состоит в том, что в организме есть две «эгоистические» системы, находящиеся иерархически выше других систем в распределении питания: центральная нервная (ЦНС) и иммунная система [23]. Когда ЦНС нуждается в повышенном количестве энергии, она может запускать процессы развития ИР метаболически зависимых тканей за счет гиперактивации паравентрикулярного ядра (ПВЯ) и дугообразного ядра (ДОЯ) гипоталамуса. ПВЯ, состоящее из оси «гипоталамус – гипофиз – надпочечники» и симпатической нервной системы, является центральным ядром стрессовых систем. Активация этого ядра подавляет захват глюкозы инсулин-чувствительными тканями, снижает высвобождение инсулина β-клетками ПЖЖ, приводя к развитию ИР. Эту ситуацию ухудшает гиперактивность ДОЯ, которое отвечает за повышение агути-родственного пептида, увеличивающего аппетит, снижающего чувствительность клеток к инсулину, предотвращающего поглощение глюкозы бурой жировой тканью и замедляющего расход энергии [24]. При сверхактивации иммунной системы происходит всплеск уровня провоспалительных цитокинов и хемокинов в крови, приводящий к отграниченному от регуляции ЦНС воспалительному процессу. Это отграничение происходит за счет продукции нейромедиаторов и гормонов на периферии и приводит к независимому от АКТГ синтезу кортизола и провоспалительных цитокинов. Большинство провоспалительных факторов обладает способностью к повышению ИР тканей разнообразными путями, в том числе за счет снижения экспрессии GLUT-4. Именно по причине первостепенности распределения энергии повышенная активность ЦНС и иммунной системы часто сопровождается болезненным состоянием и анорексией, что еще сильнее усугубляет дефицит энергии [23].
Подтверждением существования данной гипотезы является систематический обзор M. Sprengell et al., в котором рассмотрены 2804 работы, доказывающие теорию о том, что ЦНС саморегулирует свое энергетическое содержание с наивысшим приоритетом. Так, при низком содержании АТФ в нейронах происходит активация симпатоадреналовой системы, которая повышает концентрацию глюкозы в крови и снижает концентрацию инсулина. За счет этого глюкоза не поступает в инсулинозависимые ткани и через GLUT-1 проходит в мозг и иммунные клетки [25]. Подводя итог гипотезы профессора Райнера Штрауба, нельзя не упомянуть о том, что чем сложнее организована система регуляции, тем легче вывести ее из строя. Так, нарушение чувствительности рецепторов, работы ядер гипоталамуса, синтеза нейротрасмиттеров, регуляции воспалительных реакций и других неизбежно приведут к развитию ИР.
Теория, влияющая на все звенья комплексной модели. Микробиотическая гипотеза
Итак, несмотря на существование множества теорий развития ИР, ни одна из них полностью не объясняет природу процесса нарушения чувствительности периферических тканей к инсулину. Наиболее универсальной и полной является теория профессора Райнера Штрауба, объединяющая в себе центральную и иммунную регуляцию чувствительности периферических тканей к инсулину. Однако комплексная интегрированная модель ИР не включает в себя пути влияния кишечной микробиоты (КМ) на развитие ИР. Стоит отметить, что КМ за счет действия своих активных метаболитов способна действовать на все звенья теории ИР профессора Райнера Штрауба: на ЦНС, воспаление, метаболически активные ткани (печеночную, мышечную и жировую). Так, например, физиологическая концентрация ацетата, серотонина, сероводорода (H2S) и вторичных желчных кислот (ВЖК) за счет своих метаболических эффектов ассоциирована со снижением риска развития таких патологических состояний, связанных с ИР, как ожирение, АГ и дислипидемия. Более того, ГПП-1 и пептид тирозин-тирозин (PYY), синтез которых активируется КМ, воздействуют на ЦНС, вызывая тем самым инсулинозависимое потребление глюкозы тканями и снижение аппетита. Также не стоит забывать о том, что активные метаболиты КМ способствуют синтезу противовоспалительных цитокинов и сохранению целостности кишечной стенки, предотвращая развитие системного слабовыраженного воспаления, являющегося триггером ИР [23].
Кишечная микробиота и ее активные метаболиты
КМ – совокупность большого количества микроорганизмов, включающая в себя бактерии, грибы, протеи, вирусы, археи, обитающие в желудочно-кишечном тракте [26].
Доминирующими представителями КМ являются бактерии. При этом большинство родов бактерий относится к двум основным типам: Firmicutes и Bacteroides [27]. Кишечный виром состоит из множества вирусов, включая эукариотические вирусы, бактериофаги и архейные вирусы [28]. Бактериофаги (семейства Siphoviridae, Myoviridae, Podoviridae и Microviridae) составляют 90% кишечного вирома, эукариотические вирусы – 10%. У здоровых людей кишечный виром демонстрирует большую гетерогенность видов и относительную внутривидовую стабильность [29]. Грибковая КМ не такая разнообразная, как бактериальная и вирусная. Основные виды грибов – это Candida и Phialemonium [26].
В организме хозяина КМ регулирует такие процессы, как метаболизм желчных кислот, аппетит, синтез витаминов K и группы В, синтез незаменимых аминокислот, жизнедеятельность кишечного эпителия, перистальтику кишечника, переваривание сложных углеводов, гомеостаз глюкозы и липидов, формирование иммунной системы организма [30, 31]. Эти функции она осуществляет благодаря своим активным метаболитам, ферментативным системам и даже частичкам собственных клеток [32–34].
К основным активным метаболитам КМ относят КЦЖК: пропионат, ацетат и бутират, которые образуются в толстой кишке в результате анаэробной бактериальной ферментации пищевых волокон и клетчатки. Также источником бутирата является коровье молоко [35].
Не менее важными метаболитами являются индол, серотонин и кинуренины, синтезируемые тремя разными путями из экзогенного триптофана. Индол образуется путем прямого преобразования триптофана КМ, которая продуцирует фермент триптофаназу. Серотонин синтезируется за счет активации триптанином энтерохромаффинных клеток слизистой кишечника. При этом триптанин – продукт метаболизма триптофана. Путь образования кинуренинов до конца не изучен, однако считается, что их синтез обусловлен активацией индоламин-2,3-диоксигеназы под воздействием провоспалительных факторов, вырабатывающихся в том числе КМ [36, 37].
Еще одним активным метаболитом КМ является H2S. Его основными поставщиками являются сульфатредуцирующие бактерии, гидролизующие сульфатсодержащие соединения (аминокислоты, содержащие метионин или цистеин) [38].
Также КМ участвует в биотрансформации первичных желчных кислот (ПЖК) до ВЖК. Это происходит с помощью трех основных путей: деконъюгации, дегидрирования и реакции декарбоксилирования. Деконъюгация происходит за счет гидролаз желчных солей, большую часть которых синтезируют Firmicutes (30%), Bacteroidetes (14,4%) и Actinobacteria (8,9%). Дегидрирование осуществляют Clostridium spp., относящиеся к Firmicutes. В процессе деконъюгации и дегидрирования образуются литохолат и дезоксихолат [33].
Основные эффекты активных метаболитов КМ перечислены в табл. 1 [39].
Таблица 1. Основные эффекты активных метаболитов КМ [39]
Активное вещество | Клинические эффекты | |
| Серотонин | 1) усиление перистальтики кишечника; 2) увеличение секреции слизи; 3) контроль барьерной функции кишечника; 4) активация липолиза и глюконеогенеза | |
Индол | 1) противовоспалительный эффект; 2) усиление барьерной функции кишечника; 3) увеличение синтеза ГПП-1; 4) образование кишечной биопленки | |
Вторичные желчные кислоты | 1) торможение глюконеогенеза; 2) увеличение синтеза ГПП-1;3) участие в метаболизме липидов: снижение синтеза холестерина и липопротеинов низкой плотности и повышение синтеза липопротеинов высокой плотности | |
| Сероводород | 1) кардиопротективные функции: активация митохондриального дыхания в условиях ишемии миокарда, вазодилатация;2) увеличение синтеза грелина;3) снижение синтеза инсулина;4) активация синтеза ГПП-1 и пептида YY;5) иммунная регуляция | |
| Короткоцепочечные жирные кислоты | Бутират | 1) энергетический субстрат для колоноцитов;2) пролиферация и дифференцировка клеток кишечника;3) контроль апоптоза клеток кишечника;4) усиление барьерной функции кишечника;5) активация кишечного глюконеогенеза;6) противовоспалительное действие |
| Пропионат | 1) энергетический субстрат для колоноцитов;2) активация секреции инсулина;3) участие в глюконеогенезе печени;4) регуляция аппетита | |
| Ацетат | 1) регуляция центрального аппетита;2) поддержание жизнедеятельности кишечных бактерий;3) метаболизм липидов | |
Влияние кишечной микробиоты на развитие инсулинорезистентности
КМ контролирует чувствительность периферических тканей к инсулину за счет своих активных метаболитов, механизма «кишечник – мозг – периферия», противовоспалительной активности и поддержания непроницаемости кишечного барьера для эндотоксинов и липополисахаридов (ЛПС) грамотрицательных бактерий. Следовательно, при дисбиозе состава КМ резко увеличивается риск развития ИР.
Роль активных метаболитов кишечной микробиоты в развитии инсулинорезистентности
Основными продуцентами КЦЖК являются Faecalibacterium prausnitzii и Eubacterium rectale [40]. Бутират, о свойствах которого известно больше всего, в просвете желудочно-кишечного тракта (ЖКТ) выполняет множество важных функций для человеческого организма. Одной из главных является мощнейший противовоспалительный эффект, осуществляемый благодаря способности подавлять активность провоспалительных макрофагов и активировать регуляторные Т-клетки в кишечнике. Также бутират поддерживает целостность кишечного эпителия, способствуя образованию плотных контактов между клетками кишечника и повышая секрецию муцина. Более того, бутират влияет на синтез инсулина и снижение аппетита посредством увеличения концентрации ГПП-1 и PYY в просвете кишечника, что подтверждается исследованием D. Zhou et al., в котором внутривенное введение бутирата натрия лабораторным животным приводило к увеличению концентрации ГПП-1 в крови и увеличению синтеза инсулина β-клетками ПЖЖ [41]. Также есть данные о том, что благодаря своим эффектам бутират может снижать риск развития ожирения. Так, в исследовании Z. Li et al. сообщается, что бутират защищает от ожирения, вызванного диетой с избыточным содержанием жиров. В 9-недельном эксперименте лабораторные животные были разделены на две группы: 1-я получала 5%-й бутират натрия с едой, 2-я его не получала. В группе, получающей бутират, было обнаружено снижение потребления пищи на 22%, а также выраженное снижение массы тела и количества белой жировой ткани по сравнению с контрольной группой [42]. Несмотря на подтверждающие данные о роли бутирата в снижении массы тела, необходимо учитывать тот факт, что бутират может метаболизироваться печенью с образованием субстратов для биосинтеза липидов [40]. Это значит, что большие концентрации КЦЖК, в частности бутирата, могут приводить к ожирению и ИР. Так, в исследовании J. de la Cuesta-Zuluaga et al. был сделан вывод, что высокие концентрации КЦЖК в стуле пациентов были ассоциированы с ожирением, повышением системного воспаления, дислипидемией, АГ и более низким разнообразием кишечных бактерий. Это исследование подтверждает теорию о том, что повышенные концентрации КЦЖК являются маркером нарушения функционирования кишечника и появления ассоциированных с данным фактором патологий [43]. Еще одним подтверждением влияния избыточной концентрации КЦЖК на развитие ИР является исследование Y. Huang et al., проведенное на лабораторных животных. В эксперименте беременные самки мышей были разделены на две группы: одни дополнительно получали бутират, другие – нет. По лабораторным показателям полученного потомства был сделан вывод о том, что нефизиологически высокий уровень бутирата во время беременности связан с риском развития углеводных нарушений у потомства в связи с более высоким индексом HOMA-IR у новорожденных по сравнению с контрольной группой [44]. Таким образом, КЦЖК играют большую роль в регуляции синтеза инсулина, контроле аппетита, обеспечении целостности кишечного эпителия. Однако не стоит забывать о том, что разные концентрации этих метаболитов по-разному влияют на баланс липидов в организме и риск развития ожирения и ИР.
Метаболические эффекты индола объясняются его влиянием на сохранение целостности кишечного эпителия, повышение экспрессии молекул адгезии, подавление воспаления и способности синтезировать инсулин через стимуляцию продукции ГПП-1 L-клетками кишечника [45, 46]. Так, в исследовании A. Abildgaard et al., в котором самцов крыс в течение 6 нед. кормили едой, обогащенной индол-3-пропионовой кислотой, было показано, что индекс HOMA-IR и уровни глюкозы в плазме натощак были значительно снижены в испытуемой группе. При этом какой-либо динамики показателей ИР и гликемии в контрольной группе выявлено не было [47]. Противовоспалительный эффект индола подтверждается в исследовании L. Ma et al., в котором обработка индолом культивируемых печеночных клеток способствовала экспрессии гена PFKFB3 – главного регуляторного гена гликолиза, а также подавлению провоспалительной активности макрофагов, зависящей от PFKFB3. Кроме того, в ходе работы было установлено, что индол уменьшает отложение жира в печени и повышает чувствительность гепатоцитов к инсулину [48]. Таким образом, снижение образования индола в кишечнике в результате дисбиоза КМ резко увеличивает риск развития ИР.
Высвобождение серотонина индуцируют C. sporogenes и Ruminococcus gnavus [36]. Данный метаболит усиливает синтез инсулина и подавляет высвобождение глюкагона на уровне ПЖЖ; в печени способствует накоплению жира, активирует липогенез и подавляет липолиз. Таким образом, серотонин можно назвать индуктором «хранения» липидов. Однако избыточное накопление липидов в метаболически активных органах – триггер развития ИР [49]. Так, в исследовании R.L. Young et al., в котором была оценена концентрация в плазме серотонина после интрадуоденального введения глюкозы пациентам с ожирением и пациентам контрольной группы, было выявлено, что более высокие концентрации серотонина отмечались среди пациентов с ожирением. Помимо этого, у пациентов с ожирением отмечалась более высокая плотность энтерохромаффинных клеток по сравнению с группой контроля, что доказывает доминирующую роль кишечного пути образования серотонина [50]. Кроме того, большие концентрации серотонина могут индуцировать развитие воспаления в кишечнике, связываясь с рецепторами на тучных клетках и макрофагах [51]. Активация воспаления на уровне энтероцитов ассоциирована с увеличением трансмиссии ЛПС в системный кровоток и развитием системного метаболического воспаления, индуцирующего развитие ИР [39].
Самой малоизученной группой метаболитов триптофана являются кинуренины. В настоящее время известно, что они являются агонистами рецепторов арилуглеводородов, активация которых вызывает окислительный стресс, повышение воспалительных факторов и преждевременное старение [52]. Это отлично продемонстрировано в исследовании E. Yu et al., в котором в течение 5 лет изучалась когорта пациентов с повышенным уровнем кинуренинов в крови. За время наблюдения у 231 из 985 пациентов развилось сердечно-сосудистое заболевание. В связи с этим было установлено, что повышенный исходный уровень кинуренинов был связан с более высоким риском развития инфаркта миокарда и сердечно-сосудистой смерти [53]. Однако значение кинуренинов в развитии ИР еще предстоит выяснить.
H2S является газотрансмиттером эндогенных сигналов у млекопитающих. Особенность H2S в том, что он обладает бимодальным механизмом действия: низкие концентрации оказывают противооксидантное, противовоспалительное и цитопротективное действие, высокие концентрации – напротив, цитотоксический и провоспалительный эффект [54]. Высокие уровни H2S на уровне ПЖЖ снижают секрецию инсулина путем уменьшения массы β-клеток [55]. Так, в исследовании L. Wu et al. у крыс Цукера с диабетом по сравнению с метаболически здоровыми животными на фоне ИР и гипергликемии наблюдались повышенный уровень H2S, нарушение высвобождения инсулина и его общее снижение в плазме крови [56]. Таким образом, стоит предположить, что резкое увеличение H2S в крови ассоциировано с дисфункцией β-клеток ПЖЖ, усугубляющейся в условиях ИР и приводящей к развитию углеводных нарушений.
ВЖК образуются из ПЖК под действием Firmicutes (30%), Bacteroidetes (14,4%) и Actinobacteria (8,9%) [33]. По данным J.B.J. Ward et al., урсодезоксихолевая кислота ослабляет высвобождение провоспалительных цитокинов и защищает от развития воспаления на уровне кишечных эпителиоцитов [57]. Более того, ВЖК обладают большим сродством с ядерными рецепторами Фарзеноида X (FXR) и мембранными рецепторами желчных кислот 5-го типа (TGR5) по сравнению с ПЖК [33]. Активация рецептора TGR5, находящегося на мембране большинства клеток человеческого организма, увеличивает синтез ГПП-1, повышает чувствительность тканей к инсулину, стимулирует расход энергии путем повышения конверсии тироксина в трийодтиронин, усиливает противовоспалительное действие путем ингибирования ядерного фактора κB в макрофагах [46]. Активация рецептора FXR в печени снижает синтез ПЖК путем ингибирования цитохрома P450 и увеличивает их окисление, в кишечнике увеличивает секрецию фактора роста фибробластов 15/19, увеличивающего расход энергии [46]. Более того, активация FXR ассоциирована с экспрессией гена инсулина [58]. Таким образом, снижение образования ВЖК в кишечнике на фоне дисбиоза КМ, вероятно, приводит к развитию ИР, дисфункции β-клеток ПЖЖ и развитию метаболических нарушений.
Роль воспаления, возникающего на фоне дисбиоза кишечной микробиоты, в развитии инсулинорезистентности
Как уже было сказано, КМ здорового человека способствует поддержанию целостности кишечного эпителия. Важность целостности кишечного барьера объясняется предотвращением трансмиссии микроорганизмов в системный кровоток и снижением риска развития системного воспаления. Такие кишечные метаболиты, как КЦЖК и индол, синтезируемые в основном грамположительными бактериями, способствуют сохранению плотных контактов между эпителиоцитами кишечника. Это предотвращает транспорт эндотоксинов и ЛПС в системный кровоток. При дисбиозе КМ снижается экспрессия белков плотных контактов и возникает дисбаланс между гибелью и регенерацией кишечных эпителиоцитов, что увеличивает проницаемость кишечного барьера и активирует развитие системного воспалительного процесса [21, 59]. Кроме этого, при дисбиозе КМ (снижение грамположительных и увеличение грамотрицательных бактерий) резко повышается кишечная концентрация молекул зонулина. Вероятно, связывание зонулина с рецепторами на плотных контактах вызывает сокращение цитоскелета колоноцитов, приводя к повышенной проницаемости кишечной стенки и развитию системного воспаления [60]. Эффекты зонулина были изучены в исследовании J.M. Moreno-Navarrete et al., в котором повышенная концентрация зонулина у мужчин с метаболическими нарушениями была ассоциирована с более высокими уровнями индекса массы тела, триглицеридов и интерлейкина 6 (ИЛ-6) в крови. Более того, в исследовании отмечалась обратная корреляция между уровнем зонулина и чувствительностью тканей к инсулину [60].
Еще одним триггером развития системного метаболического воспаления на фоне дисбиоза КМ является повышение ЛПС грамотрицательных бактерий крови. Дело в том, что после того как ЛПС проникает в системный кровоток, он связывается с клетками Купфера в печени и вызывает активацию Т-клеток и повышенный синтез провоспалительных цитокинов – ИЛ-1, ИЛ-6, ИЛ-8, фактора некроза опухоли-α и хемокинов [61]. Эти провоспалительные молекулы активируют стресс-зависимые киназы, которые, в свою очередь, фосфорилируют субстрат инсулинового рецептора, нарушая активацию внутриклеточного инсулинового каскада и резко снижая трансмиссию GLUT-4 на поверхность клеток. Формируется ИР [21, 62].
Роль разнообразия кишечной микробиоты в развитии инсулинорезистентности
Несомненно, дисбаланс грамположительных и грамотрицательных бактерий является причиной развития ИР. Это в первую очередь объясняется тем, что доминирование грамотрицательных бактерий изменяет количество активных метаболитов КМ и повышает уровень ЛПС в просвете кишечника. Действительно, в исследовании I. Medina-Vera et al. было выявлено, что у пациентов с СД2 на фоне ИР отмечалось резкое увеличение Prevotella copri (грамотрицательная бактерия) [63]. Однако грамположительные бактерии являются активаторами энергетического запасания. Это значит, что при их доминировании также будет развиваться ИР [64]. Действительно, в исследовании A. Koliada et al. было показано, что по мере увеличения индекса массы тела отмечалось повышение Firmicutes (грамположительные бактерии) и снижение Bacteroidetes (грамотрицательные бактерии) [64]. Однако, вопреки этим результатам, в исследовании H.J. Hu et al. не было обнаружено никакой разницы в отношении Firmicutes к Bacteroidetes между пациентами с СД2 и контрольной группой [65]. Дискордантные результаты наталкивают на мысль, что не только дисбаланс между грамположительными и грамотрицательными бактериями определяет риск развития ИР, но и непосредственно количественная представленность бактерий в кишечнике. Так, результаты исследования Z. Chen et al. с участием 2166 пациентов, из которых 193 имели СД2, показали, что чем выше альфа-разнообразие (видовое) КМ, тем ниже ИР, чем меньше альфа-разнообразие, тем больше риск развития ИР и СД2 [66].
Влияние отдельных штаммов бактерий на развитие инсулинорезистентности
Важно, что не только дисбаланс грамположительных и грамотрицательных бактерий и снижение альфа-разнообразия ассоциированы с развитием ИР. Имеются данные, что отдельные представители КМ также влияют на чувствительность периферических тканей к инсулину. Так, по данным I.G. Macchione et al., Akkermansia muciniphila поддерживает непроницаемость кишечного барьера, снижает синтез факторов воспаления, предотвращает развитие ожирения и, следовательно, снижает риск развития ИР [67]. Bifidobacterium lactis и Lactobacillus gasseri увеличивают экспрессию и транслокацию GLUT-4, стимулируя инсулин-опосредованное поглощение глюкозы периферическими тканями. Lactobacillus rhamnosus увеличивает уровень адипонектина в белой жировой ткани, тем самым снижая ИР [68]. Faecalibacterium prausnitzii снижает ИР и выраженность жирового гепатоза за счет активации синтеза КЦЖК и ГПП-1 [68, 69]. Roseburia участвует в синтезе бутирата, контролирует апоптоз колоноцитов, активирует кишечный глюконеогенез, обусловливает продукцию противовоспалительных цитокинов, что снижает риск развития системного метаболического воспаления, ведущего к развитию ИР [68].
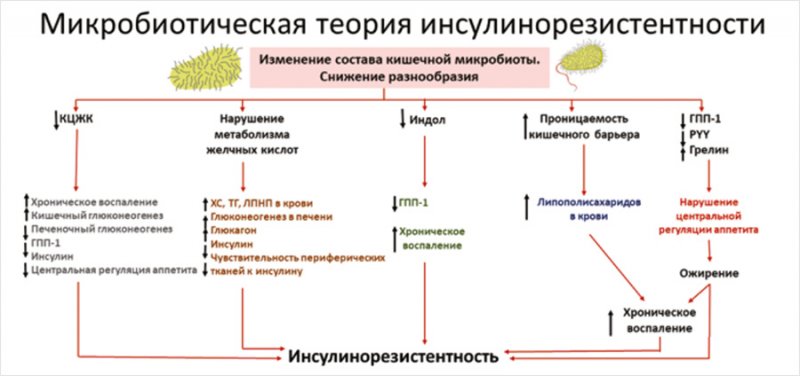
Итак, качественное и количественное изменение состава КМ может индуцировать системное воспаление и развитие ИР путем снижения или резкого повышения продукции активных метаболитов, нарушения целостности кишечного барьера, повышения молекул зонулина в крови и увеличения транспорта ЛПС в системный кровоток. Таким образом, мы можем говорить о существовании микробиотической теории развития ИР, которая представлена на рис. [39, 70]
Рисунок. Схема микробиотической теории развития инсулинорезистентности [39, 70]
Медикаментозная коррекция инсулинорезистентности метформином у пациентов с сахарным диабетом 2-го типа
Метформин является препаратом выбора в лечении СД2 уже на протяжении многих лет [71]. Его основным сахароснижающим действием считается подавление глюконеогенеза в печени, осуществляемое благодаря двум мишеням, расположенным в митохондриях гепатоцитов. К этим мишеням относятся респираторный комплекс I и митохондриальная глицерин-3-фосфатдегидрогеназа, функцию которых метформин подавляет. Это приводит к увеличению соотношения аденозинмонофосфата и аденозинтрифосфата, что приводит к подавлению экспрессии генов глюконеогенеза [72]. Кроме этого, метформин ингибирует транспорт глюкозы из кишечника в кровь путем увеличения анаэробного метаболизма глюкозы в энтероцитах [73]. Также имеются данные, что метформин способен увеличивать синтез ГПП-1 клетками кишечника и влиять на состав КМ [74].
То, что метформин действительно способен изменять состав и разнообразие КМ, доказывается рядом исследований [75–78]. Так, по данным H. Wu et al., трансплантация фекальной микробиоты мышам с углеводными нарушениями от доноров, которые в течение 4 нед. получали терапию метформином, приводила к выраженному улучшению толерантности к глюкозе [75]. В исследовании X. Tong et al. было продемонстрировало, что терапия метформином была ассоциирована с увеличением альфа-разнообразия [76]. I. Elbere et al. показали, что терапия метформином была связана со снижением численности трех родов семейства Peptostreptococcaceae через неделю после начала лечения [77], а по данным исследования M. Zhang et al. на фоне терапии метформином у мышей Цукера с диабетом повышался уровень Firmicutes, в том числе Lactobacillus, что было ассоциировано с уменьшением эндотоксемии и повышенными уровнями ГПП-1 и инсулина в крови [78]. Таким образом, метформин действительно изменяет состав КМ. Вероятно, именно поэтому внутривенное введение метформина практически не имеет терапевтического эффекта [27].
Кишечная микробиота и эффективность метформина
Известно, что эффективность метформина у разных людей неодинакова. Значит, КМ, вероятнее всего, может влиять на сахароснижающую способность метформина. На данный момент количество исследований, оценивающих влияние КМ на эффективность терапии метформином, резко ограничено, однако A. Koh et al. в своем исследовании продемонстрировали прямую связь между кишечным метаболитом пропионатом и уровнем глюкозы крови и доказали отсутствие сахароснижающего эффекта метформина в группе мышей, получающих пропионат [79]. Но по результатам данного исследования мы можем судить только о косвенном влиянии КМ на эффективность терапии метформином через призму избыточного уровня пропионата. Эффекты пропионата, других активных метаболитов КМ и отдельных кишечных представителей на действие метформина еще предстоит доказать в клинических исследованиях.
Известно, что КМ влияет на ИР путем как непосредственного воздействия активных метаболитов на ткани-мишени, так и изменения фармакодинамики метформина. Это значит, что, модулируя состав КМ, можно добиться лучших результатов в терапии СД2. Одним из возможных способов улучшения состава КМ является использование пищевых волокон, которые метаболизируются бактериями до образования в первую очередь КЦЖК [80]. Действительно, в исследовании Y. Liu et al., проведенном на лабораторных животных, было показано, что использование в течение 8 нед. экстракта полисахарида Phellinus linteus приводило к увеличению числа бактерий, продуцирующих КЦЖК [81].
Более высокий уровень активных метаболитов, образующийся на фоне терапии пищевыми волокнами, а также модуляция КМ под действием метформина может способствовать улучшению сахароснижающей способности последнего. Так, в работе J. Zheng et al. было продемонстрировано, что комбинация метформина и пребиотика маноолигосахарида способствовала улучшению толерантности к глюкозе и восстановлению островков ПЖЖ. К тому же совместное использование этих препаратов уменьшало численность условно патогенных Clostridiales и увеличивало количество Akkermansia muciniphila и Bifidobacterium pseudolongum, ассоциированных с улучшением углеводного обмена [82]. Следовательно, добавление пребиотиков к сахароснижающим препаратам, вероятно, является вполне логичной мерой, увеличивающей эффективность противодиабетических средств.
Побочные эффекты метформина, опосредованные кишечной микробиотой
У большинства людей, принимающих метформин, часто наблюдается диспепсия, развитие которой объясняется раздражением слизистой желудка и снижением всасывания глюкозы из кишечника в кровь [72]. Однако имеются данные, подтверждающие роль КМ в развитии побочных эффектов метформина. Так, I. Elbere et al. выявили прямую связь между побочными эффектами метформина и численностью Escherichia coli и Shigella spp. [77]. T. Bryrup et al. обнаружили прямую связь между бактериями семейства Sutterella, Allisonella и Akkermansia и желудочно-кишечными проявлениями на фоне терапии метформином [83]. Более того, несмотря на то что в исследовании L.J. McCreight et al. не оценивался состав КМ, авторы сделали вывод о том, что причиной развития побочных эффектов метформина являлись кишечные факторы, так как между группами пациентов, имеющих и не имеющих диспепсию, разницы между уровнями лактата, желчных кислот, серотонина и метформина в крови выявлено не было [84]. Необходимо проведение дальнейших исследований, оценивающих безопасность терапии метформином в зависимости от исходного состава КМ.
Заключение
Благодаря своим активным метаболитам КМ может изменять скорость катаболизма энергетических ресурсов человека, влиять на концентрацию ПЖК и ВЖК, контролировать целостность кишечной стенки и участвовать в развитии иммунных реакций. А значит, изменение качественного и количественного состава КМ может увеличивать риск развития ИР и заболеваний, ассоциированных с этим состоянием (СД2, АГ и др.). Препаратом первого выбора в лечении СД2 является метформин. Важно, что КМ способна влиять на эффективность и безопасность терапии данным препаратом. В настоящее время накоплено недостаточное количество данных о том, какие представители КМ ассоциированы с сахароснижающей способностью и развитием диспепсии на фоне лечения метформином. Таким образом, необходимо проведение дальнейших исследований, что в будущем позволит персонализировать подход к стартовой сахароснижающей терапии в зависимости от исходного состава КМ.
Список литературы / References
- Майоров А.Ю. Инсулинорезистентность в патогенезе сахарного диабета 2-го типа. Сахарный диабет. 2011;14(1):35-45. https://doi.org/10.14341/2072-0351-6248.
- Демидова Т.Ю., Зенина С.Г. Коррекция инсулинорезистентности - эффективный путь управления сахарным диабетом 2-го типа и другими компонентами метаболического синдрома. Лечебное дело. 2020;(2):6-15. https://doi.org/10.24411/2071-5315-2020-12206.
- Petersen M.C., Shulman G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol Rev. 2018;98(4):2133-2223. https://doi.org/10.1152/physrev.00063.2017.
- Kolb H., Kempf K., Röhling M., Martin S. Insulin: too much of a good thing is bad. BMC Med. 2020;18(1):224. https://doi.org/10.1186/s12916-020-01688-6.
- Elsayed A.K., Vimalraj S., Nandakumar M., Abdelalim E.M. Insulin resistance in diabetes: The promise of using induced pluripotent stem cell technology. World J Stem Cells. 2021;13(3):221-235. https://doi.org/10.4252/wjsc.v13.i3.221.
- Artunc F., Schleicher E., Weigert C., Fritsche A., Stefan N., Häring H.U. The impact of insulin resistance on the kidney and vasculature. Nat Rev Nephrol. 2016;12(12):721-737. https://doi.org/10.1038/nrneph.2016.145.
- Gosling A.L., Buckley H.R., Matisoo-Smith E., Merriman T.R. Pacific Populations, Metabolic Disease and ‘Just-So Stories’: A Critique of the ‘Thrifty Genotype’ Hypothesis in Oceania. Ann Hum Genet. 2015;79(6):470-480. https://doi.org/10.1111/ahg.12132.
- Hegele R.A., Cao H., Harris S.B., Hanley A.J., Zinman B. The hepatic nuclear factor-1alpha G319S variant is associated with early-onset type 2 diabetes in Canadian Oji-Cree. J Clin Endocrinol Metab. 1999;84(3):1077-1082. https://doi.org/10.1210/jcem.84.3.5528.
- Hay T. Commentary: The Invention of Aboriginal Diabetes: The Role of the Thrifty Gene Hypothesis in Canadian Health Care Provision. Ethn Dis. 2018;28(1 Suppl.):247-252. https://doi.org/10.18865/ed.28.S1.247.
- Ayub Q., Moutsianas L., Chen Y., Panoutsopoulou K., Colonna V., Pagani L. et al. Revisiting the thrifty gene hypothesis via 65 loci associated with susceptibility to type 2 diabetes. Am J Hum Genet. 2014;94(2):176-185. https://doi.org/10.1016/j.ajhg.2013.12.010.
- Speakman J.R., Westerterp K.R. A mathematical model of weight loss under total starvation: evidence against the thrifty-gene hypothesis. Dis Model Mech. 2013;6(1):236-251. https://doi.org/10.1242/dmm.010009.
- Priante E., Verlato G., Giordano G., Stocchero M., Visentin S., Mardegan V., Baraldi E. Intrauterine Growth Restriction: New Insight from the Metabolomic Approach. Metabolites. 2019;9(11):267. https://doi.org/10.3390/metabo9110267.
- Hales C.N. Fetal and infant growth and impaired glucose tolerance in adulthood: the “thrifty phenotype” hypothesis revisited. Acta Paediatr Suppl. 1997;422:73-77. https://doi.org/10.1111/j.1651-2227.1997.tb18350.x.
- Nakano Y. Adult-Onset Diseases in Low Birth Weight Infants: Association with Adipose Tissue Maldevelopment. J Atheroscler Thromb. 2020;27(5):397-405. https://doi.org/10.5551/jat.RV17039.
- Guarnotta V., Amato M.C., Pivonello R., Arnaldi G., Ciresi A., Trementino L. et al. The degree of urinary hypercortisolism is not correlated with the severity of Cushing’s syndrome. Endocrine. 2017;55:564-572. https://doi.org/10.1007/s12020-016-0914-9.
- Joseph J.J., Golden S.H. Cortisol dysregulation: the bidirectional link between stress, depression, and type 2 diabetes mellitus. Ann N Y Acad Sci. 2017;1391(1):20-34. https://doi.org/10.1111/nyas.13217.
- Пашенцева А.В., Вербовой А.Ф., Шаронова Л.А. Инсулинорезистентность в терапевтической клинике. Ожирение и метаболизм. 2017;14(2):9-17. https://doi.org/10.14341/omet201729-17.
- Steptoe A., Hackett R.A., Lazzarino A.I., Bostock S., La Marca R., Carvalho L.A., Hamer M. Disruption of multisystem responses to stress in type 2 diabetes: investigating the dynamics of allostatic load. Proc Natl Acad Sci USA. 2014;111(44):15693-15698. https://doi.org/10.1073/pnas.1410401111.
- Gadgil M.D., Appel L.J., Yeung E., Anderson C.A., Sacks F.M., Miller E.R. The effects of carbohydrate, unsaturated fat, and protein intake on measures of insulin sensitivity: results from the OmniHeart trial. Diabetes Care. 2013;36(5):1132-1137. https://doi.org/10.2337/dc12-0869.
- Mirabelli M., Chiefari E., Arcidiacono B., Corigliano D.M., Brunetti F.S., Maggisano V. et al. Mediterranean Diet Nutrients to Turn the Tide against Insulin Resistance and Related Diseases. Nutrients. 2020;12(4):1066. https://doi.org/10.3390/nu12041066.
- Scheithauer T.P.M., Rampanelli E., Nieuwdorp M., Vallance B.A., Verchere C.B., van Raalte D.H., Herrema H. Gut Microbiota as a Trigger for Metabolic Inflammation in Obesity and Type 2 Diabetes. Front Immunol. 2020;11:571731. https://doi.org/10.3389/fimmu.2020.571731.
- Iglesias Molli A.E., Penas Steinhardt A., López A.P., González C.D., Vilariño J., Frechtel G.D., Cerrone G.E. Metabolically healthy obese individuals present similar chronic inflammation level but less insulin-resistance than obese individuals with metabolic syndrome. PLoS ONE. 2017;12(12):e0190528. https://doi.org/10.1371/journal.pone.0190528.
- Straub R.H. Insulin resistance, selfish brain, and selfish immune system: an evolutionarily positively selected program used in chronic inflammatory diseases. Arthritis Res Ther. 2014;16(2 Suppl.):S4. https://doi.org/10.1186/ar4688.
- Ruud J., Steculorum S.M., Brüning J.C. Neuronal control of peripheral insulin sensitivity and glucose metabolism. Nat Commun. 2017;8:15259. https://doi.org/10.1038/ncomms15259.
- Sprengell M., Kubera B., Peters A. Brain More Resistant to Energy Restriction Than Body: A Systematic Review. Front Neurosci. 2021;15:639617. https://doi.org/10.3389/fnins.2021.639617
- Piewngam P., De Mets F., Otto M. Intestinal microbiota: The hidden gems in the gut? Asian Pac J Allergy Immunol. 2020;38(4):215-224. https://doi.org/10.12932/AP-020720-0897.
- Salazar J., Angarita L., Morillo V., Navarro C., Martínez M. S., Chacín M. et al. Microbiota and Diabetes Mellitus: Role of Lipid Mediators. Nutrients. 2020;12(10):3039. https://doi.org/10.3390/nu12103039.
- Sekirov I., Russell S.L., Antunes L.C., Finlay B.B. Gut microbiota in health and disease. Physiol Rev. 2010;90(3):859-904. https://doi.org/10.1152/physrev.00045.2009.
- Hsu C.L., Duan Y., Fouts D.E., Schnabl B. Intestinal virome and therapeutic potential of bacteriophages in liver disease. J Hepatol. 2021;75(6):1465-1475. https://doi.org/10.1016/j.jhep.2021.08.00330.
- Thursby E., Juge N. Introduction to the human gut microbiota. Biochem J. 2017;474(11):1823-1836. https://doi.org/10.1042/BCJ20160510.
- Rowland I., Gibson G., Heinken A., Scott K., Swann J., Thiele I., Tuohy K. Gut microbiota functions: metabolism of nutrients and other food components. Eur J Nutr. 2018;57(1):1-24. https://doi.org/10.1007/s00394-017-1445-8.
- Schoeler M., Caesar R. Dietary lipids, gut microbiota and lipid metabolism. Rev Endocr Metab Disord. 2019;20(4):461-472. https://doi.org/10.1007/s11154-019-09512-0.
- Winston J.A., Theriot C.M. Diversification of host bile acids by members of the gut microbiota. Gut Microbes. 2020;11(2):158-171. https://doi.org/10.1080/19490976.2019.1674124.
- Stojanović O., Trajkovski M. Microbiota guides insulin trafficking in beta cells. Cell Res. 2019;29(8):603-604. https://doi.org/10.1038/s41422-019-0200-5.
- Silva Y.P., Bernardi A., Frozza R.L. The Role of Short-Chain Fatty Acids From Gut Microbiota in Gut-Brain Communication. Front Endocrinol (Lausanne). 2020;11:25. https://doi.org/10.3389/fendo.2020.00025.
- Roager H.M., Licht T.R. Microbial tryptophan catabolites in health and disease. Nat Commun. 2018;9(1):3294. https://doi.org/10.1038/s41467-01805470-4.
- Schwarcz R., Stone T.W. The kynurenine pathway and the brain: Challenges, controversies and promises. Neuropharmacology. 2017;112(Pt B):237-247. https://doi.org/10.1016/j.neuropharm.2016.08.003.
- Barton L.L., Ritz N.L., Fauque G.D., Lin H.C. Sulfur Cycling and the Intestinal Microbiome. Dig Dis Sci. 2017;62(9):2241-2257. https://doi.org/10.1007/s10620-017-4689-5.
- Демидова Т.Ю., Лобанова К.Г., Ойноткинова О.Ш. Кишечная микробиота как фактор риска развития ожирения и сахарного диабета 2-го типа. Терапевтический архив. 2020;92(10):97-104. https://doi.org/10.26442/00403660.2020.10.000778.
- Liu H., Wang J., He T., Becker S., Zhang G., Li D., Ma X. Butyrate: A DoubleEdged Sword for Health? Adv Nutr. 2018;9(1):21-29. https://doi.org/10.1093/advances/nmx009.
- Zhou D., Chen Y.W., Zhao Z.H., Yang R.X., Xin F.Z., Liu X.L. et al. Sodium butyrate reduces high-fat diet-induced non-alcoholic steatohepatitis through upregulation of hepatic GLP-1R expression. Exp Mol Med. 2018;50(12):1-12. https://doi.org/10.1038/s12276-018-0183-1.
- Li Z., Yi C.X., Katiraei S., Kooijman S., Zhou E., Chung C.K. et al. Butyrate reduces appetite and activates brown adipose tissue via the gut-brain neural circuit. Gut. 2018;67(7):1269-1279. https://doi.org/10.1136/gut-jnl-2017-314050.
- De la Cuesta-Zuluaga J., Mueller N.T., Álvarez-Quintero R., Velásquez-Mejía E.P., Sierra J.A., Corrales-Agudelo V. et al. Higher Fecal Short-Chain Fatty Acid Levels Are Associated with Gut Microbiome Dysbiosis, Obesity, Hypertension and Cardiometabolic Disease Risk Factors. Nutrients. 2018;11(1):51. https://doi.org/10.3390/nu11010051.
- Huang Y., Gao S., Chen J., Albrecht E., Zhao R., Yang X. Maternal butyrate supplementation induces insulin resistance associated with enhanced intramuscular fat deposition in the offspring. Oncotarget. 2017;8(8):13073-13084. https://doi.org/10.18632/oncotarget.14375
- Ji Y., Gao Y., Chen H., Yin Y., Zhang W. Indole-3-Acetic Acid Alleviates Nonalcoholic Fatty Liver Disease in Mice via Attenuation of Hepatic Lipogenesis, and Oxidative and Inflammatory Stress. Nutrients. 2019;11(9):2062. https://doi.org/10.3390/nu11092062.
- Chen J., Vitetta L. Gut Microbiota Metabolites in NAFLD Pathogenesis and Therapeutic Implications. Int J Mol Sci. 2020;21(15):5214. https://doi.org/10.3390/ijms21155214.
- Abildgaard A., Elfving B., Hokland M., Wegener G., Lund S. The microbial metabolite indole-3-propionic acid improves glucose metabolism in rats, but does not affect behaviour. Arch Physiol Biochem. 2018;124(4):306-312. https://doi.org/10.1080/13813455.2017.1398262.
- Ma L., Li H., Hu J., Zheng J., Zhou J., Botchlett R. et al. Indole Alleviates Diet-Induced Hepatic Steatosis and Inflammation in a Manner Involving Myeloid Cell 6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase 3. Hepatology. 2020;72(4):1191-1203. https://doi.org/10.1002/hep.31115.
- Yabut J.M., Crane J.D., Green A.E., Keating D.J., Khan W.I., Steinberg G.R. Emerging Roles for Serotonin in Regulating Metabolism: New Implications for an Ancient Molecule. Endocr Rev. https://doi.org/https://doi.org/10.1210/er.2018-00283.
- Young R.L., Lumsden A.L., Martin A.M., Schober G., Pezos N., Thazhath S.S. et al. Augmented capacity for peripheral serotonin release in human obesity. Int J Obes (Lond). 2018;42(11):1880-1889. https://doi.org/10.1038/s41366-018-0047-8.
- Mishima Y., Ishihara S. Enteric Microbiota-Mediated Serotonergic Signaling in Pathogenesis of Irritable Bowel Syndrome. Int J Mol Sci. 2021;22(19):10235. https://doi.org/10.3390/ijms221910235.
- Kaiser H., Parker E., Hamrick M.W. Kynurenine signaling through the aryl hydrocarbon receptor: Implications for aging and healthspan. Exp Gerontol. 2020;130:110797. https://doi.org/10.1016/j.exger.2019.110797.
- Yu E., Ruiz-Canela M., Guasch-Ferré M., Zheng Y., Toledo E., Clish C. et al. Increases in Plasma Tryptophan Are Inversely Associated with Incident Cardiovascular Disease in the Prevención con Dieta Mediterránea (PREDIMED) Study. J Nutr. 2017;147(3):314-322. https://doi.org/10.3945/jn.116.2417.
- Dilek N., Papapetropoulos A., Toliver-Kinsky T., Szabo C. Hydrogen sulfide: An endogenous regulator of the immune system. Pharmacol Res. 2020;161:105119. https://doi.org/10.1016/j.phrs.2020.105119.
- Zhang H., Huang Y., Chen S., Tang C., Wang G., Du J., Jin H. Hydrogen sulfide regulates insulin secretion and insulin resistance in diabetes mellitus, a new promising target for diabetes mellitus treatment? A review. J Adv Res. 2020;27:19-30. https://doi.org/10.1016/j.jare.2020.02.013.
- Wu L., Yang W., Jia X., Yang G., Duridanova D., Cao K., Wang R. Pancreatic islet overproduction of H2S and suppressed insulin release in Zucker diabetic rats. Lab Invest. 2009;89(1):59-67. https://doi.org/10.1038/labinvest.2008.109
- Ward J.B.J., Lajczak N.K., Kelly O.B., O’Dwyer A.M., Giddam A.K., Gabhann J.N. et al. Ursodeoxycholic acid and lithocholic acid exert anti-inflammatory actions in the colon. Am J Physiol Gastrointest Liver Physiol. 2017;312(6):G550-G558. https://doi.org/10.1152/ajpgi.00256.2016.
- Ma Q., Li Y., Li P., Wang M., Wang J., Tang Z. et al. Research progress in the relationship between type 2 diabetes mellitus and intestinal flora. Biomed Pharmacother. 2019;117:109138. https://doi.org/10.1016/j.biopha.2019.109138.
- Duttaroy A.K. Role of Gut Microbiota and Their Metabolites on Atherosclerosis, Hypertension and Human Blood Platelet Function: A Review. Nutrients. 2021;13(1):144. https://doi.org/10.3390/nu13010144.
- Moreno-Navarrete J.M., Sabater M., Ortega F., Ricart W., Fernández-Real J.M. Circulating zonulin, a marker of intestinal permeability, is increased in association with obesity-associated insulin resistance. PLoS ONE. 2012;7(5):e37160. https://doi.org/10.1371/journal.pone.0037160.
- Успенский Ю.П., Барышникова Н.В., Балукова Е.В. Дисбиоз кишечника, повышение проницаемости кишечной стенки и неалкогольная жировая болезнь печени. Медицинский алфавит. 2019;4(38):48-53. https://doi.org/10.33667/2078-5631-2019-4-38(413)-48-53.
- Дедов И.И., Ткачук В.А., Гусев Н.Б., Ширинский В.П., Воротников А.В., Кочегура Т.Н. и др. Сахарный диабет 2-го типа и метаболический синдром: молекулярные механизмы, ключевые сигнальные пути и определение биомишеней для новых лекарственных средств. Сахарный диабет. 2018;21(5):364-375. https://doi.org/10.14341/DM9730.
- Medina-Vera I., Sanchez-Tapia M., Noriega-López L., Granados-Portillo O., Guevara-Cruz M., Flores-López A. et al. A dietary intervention with functional foods reduces metabolic endotoxaemia and attenuates biochemical abnormalities by modifying faecal microbiota in people with type 2 diabetes. Diabetes Metab. 2019;45(2):122-131. https://doi.org/10.1016/j.diabet.2018.09.004.
- Koliada A., Syzenko G., Moseiko V., Budovska L., Puchkov K., Perederiy V. et al. Association between body mass index and Firmicutes/Bacteroidetes ratio in an adult Ukrainian population. BMC Microbiol. 2017;17(1):120. https://doi.org/10.1186/s12866-017-1027-1.
- Hu H.J., Park S.G., Jang H.B., Choi M.K., Park K.H., Kang J.H. et al. Obesity Alters the Microbial Community Profile in Korean Adolescents. PLoS ONE. 2015;10(7):e0134333. https://doi.org/10.1371/journal.pone.0134333.
- Chen Z., Radjabzadeh D., Chen L., Kurilshikov A., Kavousi M., Ahmadizar F. et al. Association of Insulin Resistance and Type 2 Diabetes With Gut Microbial Diversity: A Microbiome-Wide Analysis From Population Studies. JAMA Netw Open. 2021;4(7):e2118811. https://doi.org/10.1001/jamanet-workopen.2021.18811.
- Macchione I.G., Lopetuso L.R., Ianiro G., Napoli M., Gibiino G., Rizzatti G. et al. Akkermansia muciniphila: key player in metabolic and gastrointestinal disorders. Eur Rev Med Pharmacol Sci. 2019;23(18):8075-8083. https://doi.org/10.26355/eurrev_201909_19024.
- Gurung M., Li Z., You H., Rodrigues R., Jump D.B., Morgun A., Shulzhenko N. Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine. 2020;51:102590. https://doi.org/10.1016/j.ebiom.2019.11.051.
- Gu Y., Wang X., Li J., Zhang Y., Zhong H., Liu R. et al. Analyses of gut microbiota and plasma bile acids enable stratification of patients for antidiabetic treatment. Nat Commun. 2017;8(1):1785. https://doi.org/10.1038/s41467-017-01682-2.
- Caricilli A.M., Saad M.J. The role of gut microbiota on insulin resistance. Nutrients. 2013;5(3):829-851. https://doi.org/10.3390/nu5030829.
- Spiering M.J. The mystery of metformin. J Biol Chem. 2019;294(17):6689-6691. https://doi.org/10.1074/jbc.CL119.008628.
- Minamii T., Nogami M., Ogawa W. Mechanisms of metformin action: In and out of the gut. J Diabetes Investig. 2018;9(4):701-703. https://doi.org/10.1111/jdi.12864.
- Rena G., Hardie D.G., Pearson E.R. The mechanisms of action of metformin. Diabetologia. 2017;60(9):1577-1585. https://doi.org/10.1007/s00125-017-4342-z.
- Apostolova N., Iannantuoni F., Gruevska A., Muntane J., Rocha M., Victor V.M. Mechanisms of action of metformin in type 2 diabetes: Effects on mitochondria and leukocyte-endothelium interactions. Redox Biol. 2020;34:101517. https://doi.org/10.1016/j.redox.2020.101517.
- Wu H., Esteve E., Tremaroli V., Khan M. T., Caesar R., Mannerås-Holm L. et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med. 2017;23(7):850-858. https://doi.org/10.1038/nm.4345.
- Tong X., Xu J., Lian F., Yu X., Zhao Y., Xu L. et al. Structural Alteration of Gut Microbiota during the Amelioration of Human Type 2 Diabetes with Hyperlipidemia by Metformin and a Traditional Chinese Herbal Formula: a Multicenter, Randomized, Open Label Clinical Trial. mBio. 2018;9(3):e02392-17. https://doi.org/10.1128/mBio.02392-17.
- Elbere I., Kalnina I., Silamikelis I., Konrade I., Zaharenko L., Sekace K. et al. Association of metformin administration with gut microbiome dysbiosis in healthy volunteers. PLoS ONE. 2018;13(9):e0204317. https://doi.org/10.1371/journal.pone.0204317.
- Zhang M., Feng R., Yang M., Qian C., Wang Z., Liu W., Ma J. Effects of metformin, acarbose, and sitagliptin monotherapy on gut microbiota in Zucker diabetic fatty rats. BMJ Open Diabetes Res Care. 2019;7(1):e000717. https://doi.org/10.1136/bmjdrc-2019-000717.
- Koh A., Mannerås-Holm L., Yunn N.O., Nilsson P.M., Ryu S.H., Molinaro A. et al. Microbial Imidazole Propionate Affects Responses to Metformin through p38γ-Dependent Inhibitory AMPK Phosphorylation. Cell Metab. 2020;32(4):643-653.e4. https://doi.org/10.1016/j.cmet.2020.07.012.
- Holscher H.D. Dietary fiber and prebiotics and the gastrointestinal microbiota. Gut Microbes. 2017;8(2):172-184. https://doi.org/10.1080/19490976.2017.1290756.
- Liu Y., Wang C., Li J., Li T., Zhang Y., Liang Y., Mei Y. Phellinus linteus polysaccharide extract improves insulin resistance by regulating gut microbiota composition. FASEB J. 2020;34(1):1065-1078. https://doi.org/10.1096/fj.201901943RR.
- Zheng J., Li H., Zhang X., Jian M., Luo C., Lu Z. et al. Prebiotic MannanOligosaccharides Augment the Hypoglycemic Effects of Metformin in Correlation with Modulating Gut Microbiota. J Agric Food Chem. 2018;66(23):5821-5831. https://doi.org/10.1021/acs.jafc.8b00829.
- Bryrup T., Thomsen C.W., Kern T., Allin K.H., Brandslund I., Jørgensen N.R. et al. Metformin-induced changes of the gut microbiota in healthy young men: results of a non-blinded, one-armed intervention study. Diabetologia. 2019;62(6):1024-1035. https://doi.org/10.1007/s00125-019-4848-7.
- McCreight L.J., Stage T.B., Connelly P., Lonergan M., Nielsen F., Prehn C. et al. Pharmacokinetics of metformin in patients with gastrointestinal intolerance. Diabetes Obes Metab. 2018;20(7):1593-1601. https://doi.org/10.1111/dom.13264.