


Врожденный гиперинсулинизм у новорожденных и детей раннего возраста: состояние проблемы и результаты хирургического лечения - «Гастроэнтерология»
Журнал "Медицинский совет" №11/2021
DOI: 10.21518/2079-701X-2021-11-226-239

Национальный медицинский исследовательский центр имени В.А. Алмазова; 197341, Россия, Санкт-Петербург, ул. Аккуратова, д. 2
Врожденный гиперинсулинизм вызывает необратимое повреждение коры головного мозга с последующей инвалидизацией детей. В статье представлены особенности этиопатогенеза, клиники заболевания. Подробно разобраны гистологические варианты поражения поджелудочной железы. Сформулированы принципы правильной постановки диагноза. Представлен новый в России метод дооперационного определения гистологической формы заболевания, который проводится в Национальном медицинском исследовательском центре имени В.А. Алмазова (НМИЦ им. В.А. Алмазова) с 2017 г., – ПЭТ/КТ (позитронноэмиссионная и компьютерная томография) с 18F-ДОФА, объяснены биохимические основы его клинического применения и методика обследования. Описаны принципы подбора медикаментозной терапии с возможными осложнениями, необходимостью адекватной оценки ее эффективности. При невозможности достигнуть стойкой целевой эугликемии без необходимости внутривенной инфузии глюкозы показана оперативная коррекция заболевания. В схематических рисунках и интраоперационных фотографиях описаны подходы к хирургическому лечению, наглядно разобраны этапы операций, возможные осложнения. Представлены результаты оперативных вмешательств в НМИЦ им. В.А. Алмазова за январь 2017 – февраль 2021 г., где было оперировано 39 детей с врожденным гиперинсулинизмом. По данным ПЭТ/КТ с 18F-ДОФА диагностированы 15 диффузных и 24 фокальные формы. После операции у 12 (31%) пациентов подтверждено диффузное поражение железы, у 23 (59%) – фокальный характер поражения, у 4 (10%) – интраоперационно диагностирована атипичная форма. Из 39 детей у 36 (92%) – полное купирование гиперинсулинизма, значительное улучшение психомоторного развития, из них 9 (23%) нуждаются в заместительной инсулинотерапии минимальными дозировками, – это 8 детей с диффузной формой заболевания и 1 ребенок – с атипичной. Интраи послеоперационных осложнений не отмечалось. Таким образом, частичная панкреатэктомия при фокальных, субтотальная при атипичных и почти тотальная при диффузных формах позволяют справиться с гипогликемиями вследствие врожденного гиперинсулинизма и предотвратить поражение центральной нервной системы новорожденных и младенцев.
Для цитирования: Сухоцкая А.А., Баиров В.Г., Никитина И.Л., Митрофанова Л.Б., Перминова А.А., Рыжкова Д.В. Врожденный гиперинсулинизм у новорожденных и детей раннего возраста: состояние проблемы и результаты хирургического лечения. Медицинский Совет. 2021;(11):226-239. https://doi.org/10.21518/2079-701X-2021-11-226-239
Конфликт интересов: авторы заявляют об отсутствии конфликта интересов.
Congenital hyperinsulinism in newborns and young children: the state of the problem and the results of surgical treatment
Almazov National Medical Research Centre; 2, Akkuratov St., St Petersburg, 197341, Russia
Congenital hyperinsulinism causes irreversible damage to the cerebral cortex with subsequent disability in children. The article presents the features of etiopathogenesis, clinical picture of the disease. The histological variants of pancreatic lesions are analyzed in detail. The principles of correct diagnosis are formulated. A new in Russia method for the preoperative determination of the histological form of the disease, which is carried out at the y, Almazov National Medical Research Centre since 2017, – PET/ CT with 18F-DOPA, explained the biochemical basis of its clinical application and the examination technique. The principles of the selection of drug therapy with possible complications, the need for an adequate assessment of its effectiveness are described. If it is impossible to achieve a stable target euglycemia without the need for intravenous glucose infusion, surgical correction of the disease is indicated. In schematic drawings and intraoperative photographs, approaches to surgical treatment are described, the stages of operations and possible complications are clearly disassembled. The results of surgical interventions at the N.N. V.A. Almazov for 01.2017–02.2021, where 39 children with congenital hyperinsulinism were operated on. According to PET/CT with 18F-DOPA, 15 diffuse and 24 focal forms were diagnosed. After surgery, in 12 (31%) patients, a diffuse lesion of the gland was confirmed, in 23 (59%) – a focal nature of the lesion, in 4 (10%) – an atypical form was diagnosed intraoperatively. Of 39 children, 36 (92%) have complete relief of hyperinsulinism, a significant improvement in psychomotor development, of which 9 (23%) need insulin replacement therapy with minimal dosages, these are 8 children with a diffuse form of the disease and 1 child with an atypical one. Intra- and postoperative complications were not observed. Thus, partial pancreatectomy for focal forms, subtotal for atypical and near total for diffuse forms, can cope with hypoglycemia due to congenital hyperinsulinism and prevent damage to the central nervous system of newborns and infants.
For citation: Sukhotskaya A.A., Bairov V.G., Nikitina I.L., Mitrofanova L.B., Perminova A.A., Ryzhkova D.V. Congenital hyperinsulinism in newborns and young children: the state of the problem and the results of surgical treatment. Meditsinskiy sovet = Medical Council. 2021;(11):226-239. (In Russ.) https://doi.org/10.21518/2079-701X-2021-11-226-239
Conflict of interest: the authors declare no conflict of interest.
Введение
Персистирующая гиперинсулинемическая гипергликемия, или врожденный гиперинсулинизм (ВГИ), как это заболевание сейчас чаще называется, был описан Irvine McQuarrie в 1954 г. [1] и представляет собой гетерогенную группу различных генетических нарушений, проявляющихся рецидивирующими эпизодами гиперинсулинемической гипогликемии, которая вызывает необратимое повреждение коры головного мозга с последующей инвалидизацией таких больных [2, 3]. Другими словами, ВГИ характеризуется неадекватной секрецией инсулина β-клетками поджелудочной железы, что приводит к развитию гипогликемии. Гиперпродукция инсулина приводит к утилизации глюкозы клетками инсулинзависимых тканей и в то же время подавляет продукцию эндогенной глюкозы, свободных жирных кислот и кетоновых тел. Подобный метаболический эффект инсулина формирует биохимическую основу, лишающую пациентов с ВГИ как глюкозы, так и альтернативных источников энергии для головного мозга, увеличивая риск развития неврологических расстройств [4]. Хотя ВГИ – редкое заболевание, оно является доказанной причиной гипогликемического поражения головного мозга и задержки психомоторного развития и, таким образом, требует своевременного неотложного начала лечения (консервативного и (или) хирургического), а, соответственно, для этого необходима как можно более ранняя постановка диагноза.
Этиопатогенез
Частота заболеваемости ВГИ варьирует от 1 на 30 000– 50 000 чел. в европейской популяции до 1 на 2 500 рождений в регионах, где распространены близкородственные браки [5].
Глюкоза проникает в β-клетки путем упрощенной диффузии при участии транспортера GLUT2, где она подвергается первому этапу гликолиза при помощи фермента глюкокиназы. В результате первой реакции фосфорилирования образуется активный метаболит глюкозы – глюкозо-6-фосфат. Глюкокиназа является своего рода сенсором глюкозы в β-клетке, определяющим внутриклеточный метаболизм глюкозы в зависимости от ее внеклеточной концентрации [6, 7]. Повышение содержания глюкозы в крови приводит к активации глюкокиназы, запускающей механизмы гликолиза, в результате которых высвобождается АТФ. Активность глюкокиназы регулируется предшественником β-субъединицы пропионил-КоАкарбоксилазы и активностью фосфофруктокиназы. Последняя, в свою очередь, регулируется изменением соотношения АТФ и АДФ в клетке. Активаторами выступают АДФ и фруктозо-2,6-бифосфат, а ингибиторами – АТФ и цитрат. Стабильное соотношение цитозольного НАД+/ НАДФ в β-клетке, необходимое для адекватных процессов гликолиза, поддерживается за счет высокой активности α-глицерофосфата, выступающего переносчиком глицеральдегид-3-фосфата в митохондрии. β-клетки также характеризуются низким содержанием ЛДГ, что приводит к поступлению большей части образованного в результате гликолиза пирувата в митохондрии. В условиях низкого содержания глюкозы в клетке митохондрия восполняет свои энергозатраты путем окисления свободных жирных кислот. Поступление глюкозы в клетку ингибирует процесс окисления свободных жирных кислот, что достигается за счет высоких уровней цитрата в клетке, образующегося как результат митохондриального анаплероза. Цитрат конвертируется в малонил-КоА, являющийся ингибитором карнитин-пальмитоилтрансферазы-1 – митохондриального мембранного энзима, ответственного за транспорт длинноцепочечных ацилкоэнзимов А (ацил-КоА) из цитозоля в митохондрию. Оставаясь в цитозоле, длинноцепочечные ацил-КоА играют роль сигнальной молекулы, активируя протеинкиназу-С, ингибируя КАТР-каналы и провоцируя тем самым экзоцитоз инсулина [8–10].
АТФ-зависимые калиевые каналы играют ключевую роль в реализации глюкозо-стимулированной секреции инсулина, являясь своего рода метаболическими сенсорами β-клеток. В нестимулированных β-клетках калиевые каналы пребывают в открытом состоянии и вместе с ионами натрия, калия и АТФазой устанавливают мембранный потенциал [11]. Механизм, ответственный за открытие каналов, предполагает наличие низкого внутриклеточного соотношения АТФ к АДФ. Увеличение данного соотношения, вызванное повышением содержания глюкозы в крови, ее метаболизмом и митохондриальной активностью, приводит к закрытию АТФ-зависимых калиевых каналов, что влечет за собой деполяризацию мембраны и открытие вольтаж-зависимых Са-каналов. Увеличение концентрации внутриклеточного Са++ активирует протеинкиназы А и С, а также стимулирует экзоцитоз инсулина и его секрецию в плазму.
На сегодняшний день описано 14 генов (ABCC8, KCNJ11, GLUD1, GCK, HADH1, SLC16A1, UCP2, HNF4A и HNF1A, HK1, PGM1 и др.), мутации которых могут приводить к нарушению секреции инсулина; дальнейшее изучение генов продолжается. Ключевым в патогенезе всех моногенных форм ВГИ является наличие дисрегуляции секреции инсулина в ответ на изменение уровня гликемии.
1. Наиболее частой причиной ВГИ являются инактивирующие мутации генов KCNJ11 и ABCC8, кодирующих белки, составляющие К+-каналы β-клеток. Мутации в данных генах выявляются в ~50% случаев ВГИ [12]. Описаны как аутосомно-рецессивные, так и аутосомно-доминантные мутации указанных генов. Существует несколько механизмов, которые могут приводить к нарушению функции каналов. Исходя из механизма, нарушающего функцию белка, мутации в генах KCNJ11 и ABCC8 принято разделять на 2 типа. К первому типу относятся мутации, приводящие к отсутствию экспрессии каналов на мембране клетки, что может быть ассоциировано с нарушением формирования или транспорта канала на мембрану [12]. Ко второму типу относят мутации, в результате которых образуются неактивные калиевые каналы, что связано с их нечувствительностью к АДФ [12]. Рецессивные гомозиготные и гетерозиготные мутации в генах KCNJ11 и ABCC8 являются причиной наиболее тяжелых диффузных форм ВГИ, как правило, резистентных к терапии диазоксидом и зачастую требующих хирургического лечения [12]. Доминантные инактивирующие мутации, как правило, приводят к более мягкому течению ВГИ. Отличительной чертой большинства доминантных форм ВГИ является их чувствительность к диазоксиду, что объясняется сохранной экспрессией каналов на мембране клетки [13–16]. Формирование фокусов происходит в случае наследования отцовской гетерозиготной мутации в генах ABCC8 и KCNJ11 и соматической потери материнской аллели в регионе импринтинга на 11р15. При этом происходит снижение экспрессии генов H19 и Р57KIP2с материнской аллели, являющихся супрессорами опухолевого роста, и увеличивается экспрессия гена, кодирующего инсулиноподобный фактор роста-2 (IGF2) с отцовской аллели [17–19]. IGF2 является мощным фактором клеточной пролиферации [19]. Изменение соотношения экспрессии генов ведет к развитию локальной β-клеточной гиперплазии – формированию фокуса. Фокальные формы составляют около 30–40% всех случаев ВГИ и в подавляющем большинстве случаев характеризуются диазоксид-резистентным течением.
2. Мутации гена KCNQ1 кодируют порообразующий регион α-субъединицы вольтаж-зависимого калиевого канала Kv7.1. Предположительно дисрегуляция секреции инсулина при мутациях Kv7.1 является следствием нарушения реполяризации мембраны.
3. Активирующие аутосомно-доминантные мутации гена GLUD1 являются второй наиболее распространенной причиной ВГИ. Мутации этого гена выявляются в 10–15% [20, 21]. Ген GLUD1 расположен на длинном плече 10-й хромосомы и содержит 13 экзонов, кодирующих фермент глутаматдегидрогеназа (ГДД). ГДД – это митохондриальный фермент, катализирующий реакцию превращения глутамата в α-кетоглутарат и аммоний. В β-клетках α-кетоглутарат, участвуя в цикле Кребса, приводит к повышению концентрации внутриклеточной АТФ. Увеличение соотношения АТФ/АДФ влечет за собой закрытие АТФ-зависимых К+-каналов, деполяризацию мембраны и открытие вольтаж-зависимых Са++- каналов, что в свою очередь приводит к проникновению Са++ в клетку, стимулируя экзоцитоз инсулина [21]. Активность фермента ГДД регулируется сложным взаимодействием ее аллостерических активаторов и ингибиторов. Активирующим воздействием на ГДД обладают лейцин и АДФ, тогда как гуанозин-5-трифосфат (ГТФ) является ее потенциальным ингибитором [17]. Аллостерическая активация глутаминолизиса является одним из механизмов стимуляции лейцином секреции инсулина [22]. Активирующие мутации в гене GLUD1 снижают чувствительность ГДД к ее ингибитору ГТФ и в редких случаях приводят к повышению активности самой 37-ГДД [22]. Отсутствие ингибирующего влияния ГТФ в присутствии активирующего влияния лейцина влечет за собой гиперсекрецию инсулина. Течение гипогликемического синдрома, как правило, мягкое и хорошо поддается терапии диазоксидом в комбинации с низкопротеиновой диетой.
4. Мутации в гене HADH. Митохондриальное β-окисление жирных кислот является физиологическим адаптивным механизмом восполнения энергетических затрат в период голодания, гипертермии и при физической нагрузке. Конечным продуктом окислительных реакций является ацетил-КоА. Ген HADH кодирует фермент 3-гидроксиацилКоА-дегидрогеназу. Данный фермент катализирует предпоследнюю реакцию в процессе β-окисления короткоцепочечных жирных кислот, в результате которой происходит преобразование 3-гидроксиацил-КоА в 3-кетоацил-КоА. Инактивирующие аутосомно-рецессивные мутации HADH снижают ингибирующее влияние на ГДД, что приводит к развитию гиперсекреции инсулина в ответ на стимуляцию лейцином [23–26]. Имеется значительная вариабельность течения гипогликемического синдрома: от эпизодических, легких, поздно манифестирующих гипогликемий до тяжелых случаев с неонатальным дебютом.
Кроме того, ВГИ может быть ассоциирован с такими специфическими синдромами, как Беквита – Видемана, Перлмана и Сотоса, каждый из которых подразумевает соответствующие хромосомные аномалии.
Клинические проявления
Клиническая картина ВГИ вариабельна и мало специфична. Условно симптомы можно разделить на 2 группы: ассоциированные с активацией вегетативной нервной системы (чувство голода, плаксивость, раздражительность, парестезии, тремор, тахикардия, усиленное потоотделение) и симптомы нейрогликопении (сонливость, обмякание, помрачение/спутанность сознания, судороги, развитие комы). В неонатальном периоде характерно развитие дыхательных нарушений (брадипноэ, апноэ). Учитывая неспецифичность первых симптомов гипогликемии, поводом для измерения глюкозы в крови, как правило, служит наличие явной симптоматики (судороги, потеря сознания), что существенно увеличивает риски неврологических осложнений, в особенности если речь идет о неонатальной гипогликемии [27]. Это объясняет, почему целесообразно у всех новорожденных сразу после рождения исследовать уровень глюкозы.
ВГИ, как правило, манифестирует в неонатальный период, однако возможен и более поздний дебют, вплоть до трехлетнего возраста [28, 29]. Чем раньше проявляется заболевание, тем тяжелее оно протекает [28]. Гипогликемии при ВГИ обычно носят тяжелый характер и быстро приводят к развитию судорог и потере сознания. Описаны и мягкие формы, протекающие почти бессимптомно, проявляющиеся лишь гиподинамией и сниженным аппетитом [29]. Гиперинсулинизм у новорожденных порой может быть транзиторным, но в связи с этим необходимо проведение дифференциальной диагностики, поскольку вопрос о хирургическом лечении ставится только при доказанных персистирующих формах.
Гистологические варианты врожденного гиперинсулинизма
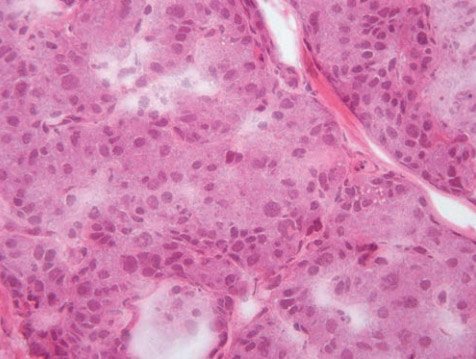
Патоморфология ВГИ неоднородна, и в настоящее время можно выделить 3 ее типа: фокальный, диффузный и атипичный [28, 29]. При фокальной форме, ассоциированной с унаследованной отцовской гетерозиготной мутацией в генах KATP и соматической утратой материнской аллели в регионе импринтинга на 11р15, поражается ограниченный участок поджелудочной железы, в котором развивается аденоматозная гиперплазия β-клеток, приводящая к слиянию островков Лангерганса (рис. 1).
Рисунок 1. Интраоперационная гистологическая картина
при фокальной форме врожденного гиперинсулинизма:
зона аденоматозной гиперплазии с увеличенными в размерах ядрами эндокриноцитов. Окрашивание гематоксилинэозином, увеличение в 400 раз
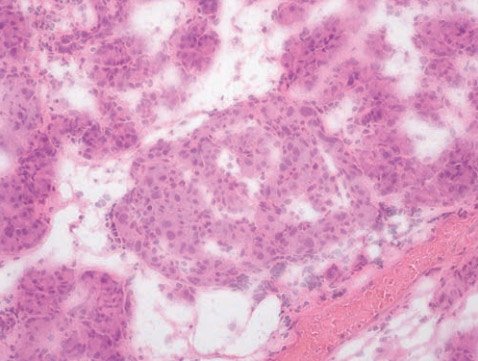
При этом, в отличие от инсулиномы, сохраняется дольчатая и островковая архитектоника с участием не-β-эндокриноцитов в пораженной области. Кроме того, при фокальном типе гиперинсулинизма возможно распространение процесса в окружающих тканях по типу щупалец осьминога. При этом у пациентов снижается экспрессия генов, являющихся супрессорами опухолевого роста, и увеличивается экспрессия гена, кодирующего фактор пролиферации, что приводит к формированию участка аденоматоза с избыточной секрецией инсулина [30]. Для диффузной формы, которая является следствием аутосомно-рецессивных мутаций, характерна гиперплазия островков Лангерганса с появлением в них гипертрофированных и гиперхромных ядер β-клеток, причем такие изменения наблюдаются во всей ткани поджелудочной железы (рис. 2). Атипичная форма отличается морфологическим мозаицизмом, включающим различные сочетания фокального и диффузного поражения железы [31]. Генетические механизмы развития атипичных форм в настоящий момент остаются неизвестными.
Рисунок 2. Интраоперационная гистологическая картина при диффузной форме: островок Лангерганса с увеличенными в размерах ядрами эндокриноцитов, окрашивание гематоксилин-эозином, увеличение в 200 раз
Диагностика врожденного гиперинсулинизма
ВГИ характеризуется гиперинсулинемической, гипокетотической гипогликемией с низким содержанием свободных жирных кислот в крови. Используются следующие диагностические критерии:
- Уровень гликемии в крови менее 2,6 ммоль/л.
- В момент гипогликемии, подтвержденной лабораторно, уровень С-пептида и (или) инсулина нормальный или повышенный (более 2 МЕ/л), отсутствуют кетоны, нет повышения свободных жирных кислот (менее 1,5 ммоль/л). Необходимо подчеркнуть, что глюкоза крови, инсулин и С-пептид должны быть взяты одновременно (в одной пробе крови).
- Положительный ответ в тесте с глюкагоном (введение 0,5 мг глюкагона вызывает прирост гликемии > 30 мг/дл).
- Вспомогательным критерием является высокая потребность в глюкозе для поддержания состояния эугликемии – более 8 мг/кг/мин.
- Подтверждение диагноза возможно и пробой с голоданием (во время гипогликемии, спровоцированной голоданием, определяются уровни инсулина и С-пептида в крови).
Предоперационная дифференциальная диагностика гистологических вариантов ВГИ очень важна, так как во многом определяет тактику лечения пациентов. При фармакорезистентном течении диффузного ВГИ частичная панкреатэктомия не всегда приводит к улучшению контроля над гликемией, поэтому большинством исследователей рекомендована субтотальная панкреатэктомия, но это сопряжено с высоким риском развития инсулинозависимого сахарного диабета. Однако хирургическое лечение в таких случаях абсолютно необходимо при неэффективности консервативной терапии для предотвращения разрушительного воздействия рецидивирующей гипогликемии на психомоторное развитие с целью предотвращения инвалидизации детей [32].
В случае фокальных форм проводится селективная резекция фокуса, которая приводит к полному выздоровлению пациента с сохранением функции оставшейся нормальной поджелудочной железы [33, 34].
Стандартные методы визуализации, такие как мультиспиральная компьютерная томография (МСКТ), магнитнорезонансная томография (МРТ) и ультразвуковое исследование (УЗИ) не позволяют локализовать патологический очаг при фокальной форме [33, 34].
До недавнего времени в клинической практике для дифференциальной диагностики диффузной и фокальной форм ВГИ использовались методики, основанные на определении уровня инсулина в пробах крови, оттекающей от поджелудочной железы: чрескожный чреспеченочный забор крови из ветвей воротной вены (ЧЧЗКВ) и забор крови из правой печеночной вены после внутриартериальной стимуляции (АСЗК) кальцием различных отделов поджелудочной железы [35–37]. Чувствительность ЧЧЗКВ и АСЗК составляет 87 и 71% соответственно [35]. Специфичность данных методов варьируется в различных литературных источниках. Точность локализации фокуса гиперинсулинизма при АСЗК – 82% (95% ДИ: 72–90%) – несколько выше, чем при проведении ЧЧЗКВ – 76% (95% ДИ: 65–85%) у пациентов с фокальной формой ВГИ [35]. Эта процедура была инвазивной, длительной по времени, и результат часто не был корректен в определении очага гиперинсулинизма.
Огромное диагностическое значение для визуализации фокального или диффузного поражения в последние годы заняла ПЭТ/КТ с 18F-ДОФА (позитронноэмиссионная и компьютерная томография), которая стала применяться с 2003 г. Впервые о применении ПЭТ/КТ с 18F-ДОФА с целью дифференциальной диагностики диффузной и фокальной форм гиперинсулинизма сообщили M.J. Riberio et al. в 2005 г. [38]. На сегодняшний день данная методика является золотым стандартом дифференциальной диагностики фокальных и диффузных форм ВГИ.
18F-фтор-L-дигидроксифенилаланин (18F-ДОФА) – синтетический аналог предшественника норадреналина – фенилаланина, меченный позитрон-излучающим изотопом 18F. В основе фармакодинамики этого радиофармацевтического препарата (РФП) лежит способность островковых клеток поджелудочной железы захватывать L-дигидроксифенилаланин с его последующим метаболическим превращением в допамин с помощью L-аминокислотной декарбоксилазы [39, 40]. Подобно своему биологическому аналогу, 18F-ДОФА транспортируется в опухолевую клетку при помощи трансмембранного переносчика аминокислот L-типа. Затем 18F-ДОФА претерпевает метаболическое превращение в 18F-фтордопамин под действием L-аминокислотной декарбоксилазы и депонируется в секреторных гранулах [41, 42].
Экспериментальные исследования на лабораторных животных показали, что, помимо центральной нервной системы, синтез и хранение допамина и L-ДОФА происходят в островках поджелудочной железы [43]. Следует отметить, что у человека β-клетки экспрессируют допаминовые рецепторы [44], с помощью которых допамин и L-ДОФА подавляют секрецию инсулина [43, 45]. В исследованиях отмечено, что в случае ВГИ активность фермента L-аминокислотной декарбоксилазы повышена. Увеличенный захват 18F-ДОФА β-клетками поджелудочной железы обеспечивает позитивную радионуклидную визуализацию патологического очага. У детей захват 18F-ДОФА здоровой тканью поджелудочной железы ниже, чем у взрослых людей, что объясняется функциональной незрелостью системы L-декарбоксилазы ароматических аминокислот, благодаря чему улучшается визуализация участка с повышенным захватом радиофармпрепарата [46].
Данные ПЭТ c 18F-ДОФА дают представление о функциональной активности β-клеток поджелудочной железы, что позволяет дифференцировать фокальную и диффузные формы ВГИ. В случае фокальной формы гиперинсулинизма аккумуляция 18F-ДОФА β-клетками гораздо интенсивнее в области очага поражения по сравнению с нормальной тканью поджелудочной железы [35, 39, 47]. При диффузной форме ВГИ захват РФП повышен во всей ткани поджелудочной железы с небольшим преобладанием активности в области головки [47–49].
Детям младшего возраста ПЭТ/КТ с 18F-ДОФА выполняют под наркозом. По различным данным для получения наиболее достоверных результатов в качестве подготовки к процедуре пациентам рекомендуется отмена терапии октреотидом и глюкагоном за 48 ч до исследования [50]. Однако некоторые авторы утверждают, что медикаментозную терапию перед процедурой отменять необязательно [38, 51]. Инъекцию 18F-ДОФА осуществляют внутривенно из расчета 4 МБк на 1 кг массы пациента [48, 51, 52]. Десятиминутные ПЭТ-сканы выполняют через 10, 20, 30, 40 и 50 мин после инъекции РФП [48, 51]. Для топической диагностики очага поражения производится автоматическое совмещение трехмерных изображений ПЭТ и КТ [50].
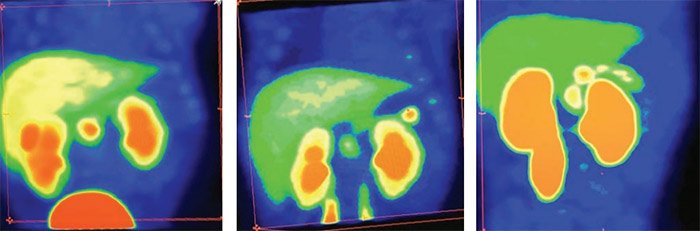
В первую очередь производится визуальная оценка ПЭТ-данных: равномерная гиперфиксация 18F-ДОФА во всех отделах поджелудочной железы характерна для диффузного ВГИ (рис. 3), а очаговое накопление РФП свидетельствует о фокальной форме заболевания (рис. 4–6).
Рисунок 3. Картина ПЭТ/КТ при диффузном распределении радиофармпрепарата, что соответствует диффузной форме врожденного гиперинсулинизма
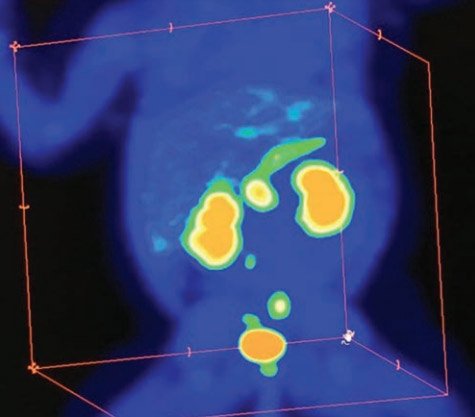
Рисунок 4. Картина поражения головки поджелудочной железы по данным ПЭТ/КТ Рисунок 5. Картина фокального поражения хвоста поджелудочной железы по данным ПЭТ/КТ
Рисунок 6. Картина поражения тела поджелудочной железы по данным ПЭТ/КТ
Для точности интерпретации результатов исследования, помимо визуальной оценки, также используются количественные показатели.
В клинической практике широко применяется стандартизированный показатель накопления (Standardized Uptake Value–SUV), который отражает концентрацию РФП в очаге, выраженную в Бк/мл к общему значению введенной в организм пациента активности РФП и нормированный на площадь поверхности тела, массу тела или безжировую массу тела пациента. Как правило, в расчет принимают максимальное значение показателя SUV – SUVmax. По мнению различных исследователей, для интерпретации результатов следует сочетать визуальную оценку с расчетом SUVmax [38, 51, 52]. У пациентов с фокальной формой ВГИ в области аденоматоза SUVmax на 50% больше, чем этот показатель в здоровой ткани поджелудочной железы [48, 52]. Для большей наглядности M. Meintjes et al. [48] рассчитали панкреатический индекс (Pancreatic Ratio, PR) как соотношение SUVmax и следующего, меньшего по значению, SUVmax в головке, теле и хвосте поджелудочной железы. Оказалось, что у пациентов с диффузной формой ВГИ панкреатический индекс не превышал 1,3, в то время как у всех детей с фокальной формой заболевания панкреатический индекс был более 1,5. До настоящего времени не было зафиксировано каких-либо осложнений после проведения ПЭТ/КТ c 18F-ДОФА у детей с ВГИ. Наибольший риск при использовании данной методики представляет ионизирующее облучение. Радиационное облучение при совмещенном использовании ПЭТ и КТ и силы тока 80 мАм/с с пиковым напряжением в 140 кВ составляет примерно от 3 до 5 мЗв [35]. В целях безопасности рекомендуется снижать дозу лучевой нагрузки до минимально эффективной. По данным литературы чувствительность ПЭТ/КТ c 18F-ДОФА при ВГИ находится в пределах 89% (95% ДИ: 81–95%), специфичность – 98% (95% ДИ: 89–100%) [53]. Подсчитано, что диагностическая точность ПЭТ/КТ c 18F-ДОФА для определения локализации аденоматоза при фокальной форме ВГИ соответствует 82% [35].
Для лучшей интерпретации исследования следует учитывать возможные причины ложноположительных результатов: физиологическую гиперфиксацию РФП головкой поджелудочной железы, различный градиент аденоматоза с большим поглощением 18F-ДОФА в области головки при диффузной форме ВГИ [54]. К ложноотрицательным результатам могут привести: малые размеры очага (менее 1 см) или мультифокальность, особенности формы аденоматоза (плоский очаг), суперпозиция левой почки, желчного пузыря, двенадцатиперстной кишки на патологический фокус [55].
Перспективной технологией исследования является совмещенная ПЭТ/МРТ с 18F-ДОФА, которая позволит существенно снизить лучевую нагрузку на пациентов с ВГИ, так как выполнение МРТ не сопряжено с воздействием на организм ребенка ионизирующего излучения [56].
До настоящего времени проведение ПЭТ/КТ с 18F-ДОФА доступно лишь в нескольких центрах в мире.
Лечение
Всем пациентам с гиперинсулинизмом показана безотлагательная медикаментозная терапия. Именно ранняя постановка диагноза и своевременное начало лечения предопределяют сохранение центральной нервной системы младенца. Такие факторы, как возраст дебюта, сопутствующие заболевания, тяжесть в сочетании с частотой и длительностью эпизодов гипогликемии предопределяют неврологический прогноз.
Ведущая задача медикаментозной терапии – достижение нормогликемии (целевой диапазон уровня глюкозы крови – 4–8 ммоль/л) путем подавления секреции инсулина. Для этого назначаются препараты, подавляющие секрецию инсулина, и (или) принимается решение о необходимости оперативного лечения, поскольку до 45–60% детей с ВГИ плохо поддаются медикаментозному лечению и требуют хирургического вмешательства [57–59].
Медикаментозная терапия
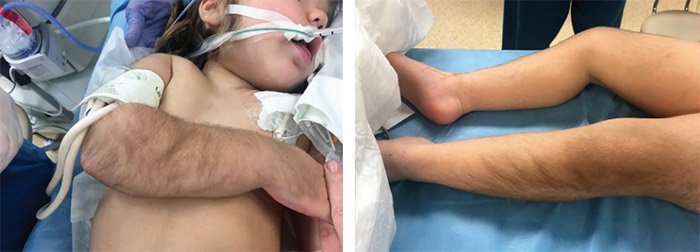
Препаратом первой линии для лечения ВГИ является диазоксид, который воздействует на К-каналы, активируя их. Тем не менее дети с диффузными формами заболевания вследствие мутаций в генах ABCC8 и KCNJ11 и большинство пациентов с фокальными формами невосприимчивы к лечению диазоксидом. Диазоксид назначается перорально в дозировке 5–15 мг/кг/сутки, разделенном на три приема. Наиболее частые побочные эффекты включают гипертрихоз, задержку жидкости и воды, что может привести к тяжелым осложнениям, таким как сердечная недостаточность и открытие артериального протока. Другие побочные эффекты диазоксида: рвота, тошнота, потеря аппетита. Диазоксид обычно назначается вместе с тиазидным диуретиком (хлоротиазидом 7–10 мг/кг/сутки, разделенном на два приема) для предотвращения задержки жидкости [61–63] (рис. 7).
Рисунок 7. Картина побочного действия диазоксида – гипертирихоза (появления избыточного роста волос как в типичныхместах, так и там, где волосы обычно не растут – лоб, щеки)
Октреотид – длительно действующий аналог соматостатина, который подавляет секрецию различных гормонов, включая инсулин, и является препаратом второй линии лечения пациентов, невосприимчивых к диазоксиду. Октреотид ингибирует секрецию инсулина, индуцируя гиперполяризацию β-клеток путем подавления кальциевых каналов. Кроме того, октреотид активирует рецепторы соматостатина (STTR 5), приводя к подавлению биосинтеза инсулина. Октреотид назначается в виде повторных подкожных инъекций 4–5 р/день или путем продолженной подкожной инфузии с использованием инсулиновой помпы в дозировке 5–35 мкг/кг/день. После первых 2–3 доз оценивается его эффективность и, возможно, требуется увеличение дозировки. Побочные эффекты включают потерю аппетита, тошноту, кишечные колики, диарею; редко встречаются серьезные побочные эффекты: медикаментозно-индуцированный гепатит, некротический энтероколит, синдром удлинения интервала QT. Кроме того, применение октреотида приводит к подавлению гормона роста, тиреотропного и адренокортикотропного гормонов [61–63].
Также в ряде случаев возможно применение следующих препаратов (их применение изучается): ланреотид (длительно действующий аналог октреотида), блокатор кальциевых каналов нифедипин, стимулятор гликогенолиза и глюконеогенеза глюкагон, антагонист глюкагоноподобного рецептора (GLP1) экзендин, иммуносупрессант сиролимус.
Хирургическое лечение
Медикаментозная терапия является терапией первой линии. Однако когда пациент не отвечает на медикаментозную терапию и не может быть отлучен от внутривенного введения глюкозы, должно быть проведено хирургическое лечение для предотвращения необратимого поражения центральной нервной системы младенца. В этих случаях дифференциальный диагноз диффузной и фокальной форм должен быть произведен дооперационно.
Первая субтотальная панкреатэктомия, проведенная у ребенка, была сообщена в 1934 г. Эвартсом Грэмом. Панкреатэктомия привела к исчезновению гипогликемии у пациента. Поджелудочная железа была исследована гистологически, но аденомы обнаружено не было [64]. В детском госпитале в Филадельфии оперативные вмешательства при ВГИ выполняются с 1950 г., достигнув в настоящее время наибольшего опыта в мире – более 500 операций (ведущий хирург – N. Scott Adzick) [65].
В Национальном медицинском исследовательском центре имени В.А. Алмазова оперативное лечение детей с ВГИ проводится с 2011 г. С 2017 г. в предоперационное обследование введены ПЭТ/КТ с 18F-ДОФА и интраоперационно экспресс-биопсия поджелудочной железы. В предоперационном периоде больной госпитализируется за сутки в отделение детской хирургии или реанимации (в зависимости от тяжести состояния); продолжается частое дробное кормление каждые 3 ч, инсулиностатическая терапия (целевая гликемия – 4–8 ммоль/л); в день операции с 6:00 утра – голод, отмена инсулиностатической терапии, титрование нагрузки глюкозой стартово 8–9 мг/кг/мин, целевая гликемия – 4–8 ммоль/л. Проводится контроль гликемии каждые 30 мин с коррекцией углеводной нагрузки.
Доступ при открытых операциях осуществляется посредством поперечной супраумбиликальной лапаротомии. Также в настоящее время можно проводить оперативное лечение путем лапароскопии. Недостатком лапароскопического подхода является слабая тактильная обратная связь, помогающая определить местонахождение невидимого очагового поражения.
Поджелудочная железа обнажается входом в полость малого сальника путем мобилизации желудочноободочной связки. Мобилизация селезенки требуется крайне редко.
Пациентам с подозрением на диффузный ВГИ проводят интраоперационную биопсию для подтверждения диагноза. Для этого берут множественные отдельные биопсии диаметром 2–3 мм из головки, тела и хвоста поджелудочной железы. После гистологического подтверждения диффузного поражения железы проводят субтотальную (95%) или почти полную панкреатэктомию (98–99%). Субтотальная панкреатэктомия (95%) включает резекцию всей поджелудочной железы от хвоста до головки, включая удаление крючковидного отростка, оставляя только крошечный участок железы между общим желчным протоком и внутренней частью подковы двенадцатиперстной кишки (рис. 8, 9).
Рисунок 8. Схема субтотальной резекции поджелудочной
железы в объеме 95% (иллюстрации выполнена художником Ю.М. Проценко по эскизам авторов)
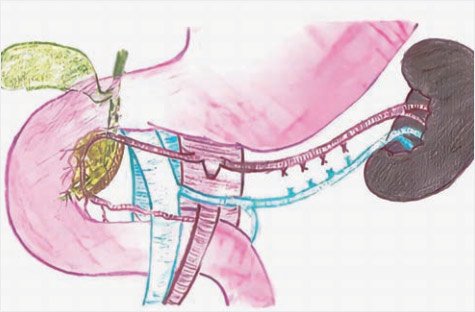
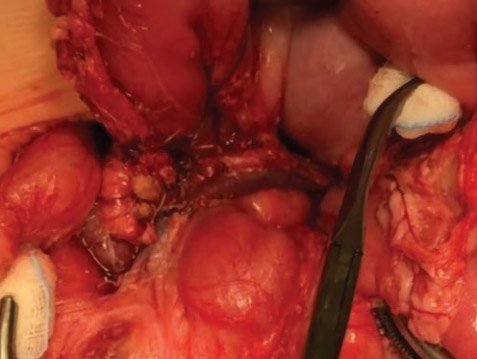
Рисунок 9. Интраоперационная картина субтотальной
резекции поджелудочной железы в объеме 95%
Для выполнения почти полной панкреатэктомии (98–99%) дополнительно удаляется участок железы между общим желчным протоком и внутренней частью подковы двенадцатиперстной кишки. Для этого полностью выделяется внутрипанкреатический ход общего желчного протока и оставляется лишь крошечный участок в области панкреатодуоденальных сосудов при панкреатэктомии (98%) (рис. 10) и не оставляется видимой ткани даже при панкреатэктомии (99%) (рис. 11, 12).
Рисунок 10. Схема субтотальной резекции поджелудочной
железы в объеме 98% (иллюстрации выполнена художником Ю.М. Проценко по эскизам авторов)
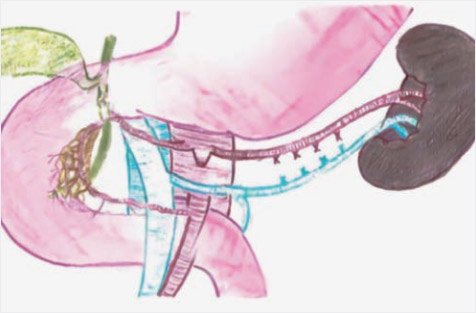
Рисунок 11. Схема субтотальной резекции поджелудочной
железы в объеме 99% (иллюстрации выполнена художником Ю.М. Проценко по эскизам авторов)
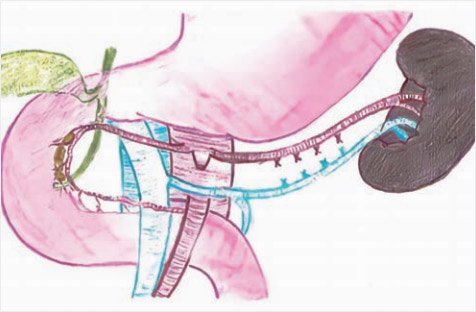
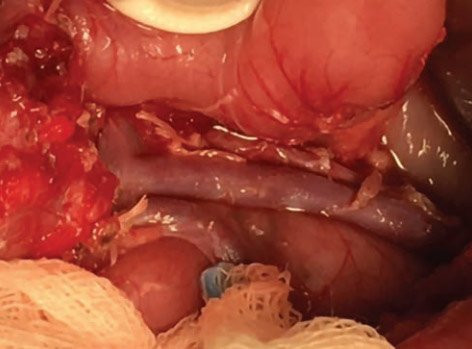
Рисунок 12. Интраоперационная картина почти тотальной
резекции поджелудочной железы в объеме 99%
При предположительно фокальной форме гиперинсулинизма по данным генетического обследования и ПЭТ/ КТ после раскрытия полости малого сальника поджелудочную железу тщательно осматривают и пальпируют, чтобы попытаться визуализировать очаговое поражение. В ряде случаев очаги определяются визуально в виде округлых дополнительных выступов (рис. 13, 14), при этом, в отличие от инсулином, они не имеют плотной капсулы и сохраняют визуально дольчатую структуру поджелудочной железы; другие определяются в виде плотных на ощупь участков в толще железы; третьи ни визуально, ни пальпаторно определить не представляется возможным.
Рисунок 13. Интраоперационная картина: фокусы аденоматозной гиперплазии видны визуально на задней поверхности головки железы, справа – в области тела
Рисунок 14. Интраоперационная картина: фокусы аденоматозной гиперплазии видны визуально в области тела
железы
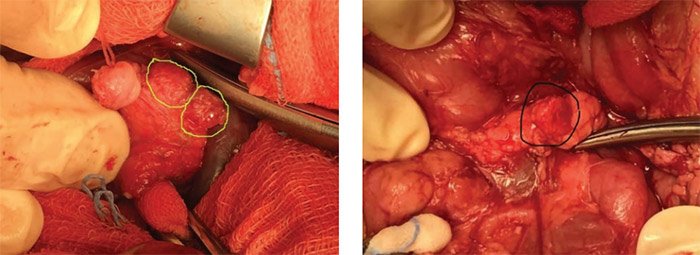
Если очаговое поражение не обнаружено, берут множественные отдельные биопсии диаметром 2–3 мм из головки, тела и хвоста поджелудочной железы. Когда биопсия демонстрирует нормальную гистологию поджелудочной железы, проводится дальнейший поиск очагового поражения с использованием данных дооперационной локализации. Дополнительные биопсии подозрительных участков производятся до тех пор, пока очаговое поражение не будет подтверждено данными экспресс-биопсии замороженных срезов. Экспертная патоморфологическая интерпретация данных экспрессбиопсии в данный момент определяет дальнейшую тактику хирурга. Очаговые поражения обычно имеют размер от 3 до 15 мм (хотя они могут быть намного больше) и часто имеют неправильную форму. У некоторых очагов есть поражение подлежащей ткани железы по типу щупалец осьминога, что делает обязательным интраоперационное подтверждение четких границ патологической и здоровой ткани срочным гистологическим исследованием замороженных срезов. Точные предоперационные исследования локализации также значительно облегчают визуальный поиск очагового поражения. В некоторых случаях поражение будет более плотным, чем окружающая нормальная поджелудочная железа, однако крошечное очаговое поражение может быть скрыто внутри поджелудочной железы, и его невозможно увидеть или почувствовать. В более успешной интраоперационной визуализации или пальпации очагового поражения, безусловно, играет роль опыт оперирующего хирурга.
После выявления очагового поражения выполняется частичная резекция поджелудочной железы и обязательно проводится срочный гистологический анализ краев для обеспечения полной резекции (чтобы не пропустить наличие поражения ткани по типу щупалец осьминога в остающейся ткани железы). При поверхностном расположении очагов возможна их энуклеация. Однако следует помнить, что для достоверности и адекватности резекции необходима экспресс-биопсия подлежащей ткани по всей полуокружности очага, а при выявлении поражения по типу щупалец осьминога показано удаление и этой ткани. Кроме того, у новорожденных и детей раннего возраста визуализировать проток поджелудочной железы практически не представляется возможным, поэтому манипуляции в глубине ткани железы очень опасны его повреждением. По этим причинам при поражении тела и хвоста обычно выполняется дистальная резекция поджелудочной железы. При поражении тела ближе к перешейку железы целесообразно сохранение дистального здорового участка железы путем панкреатоеюностомии.
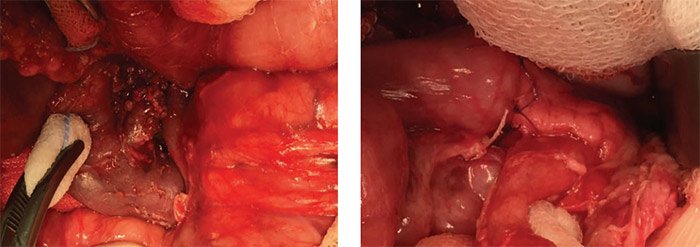
При поражении головки поджелудочной железы рядом с общим желчным протоком или протоком поджелудочной железы может быть сложно иссечь поражение целиком, особенно если есть щупальца пораженной ткани, которые отходят от очага в глубжележащие ткани. Чтобы обеспечить полную резекцию поражения в этих сложных случаях, целесообразно удаление большей части или почти всей головки поджелудочной железы (рис. 15) с последующей панкреатикоеюностомией по Ру для дренирования оставшегося тела и хвоста поджелудочной железы. У младенцев проток поджелудочной железы на поверхности разреза перерезанного тела поджелудочной железы не виден, поэтому между концом тощей кишки по Ру и капсулой тела поджелудочной железы создается тщательный анастомоз отдельными узловыми швами викрилом 6/0 для эффективного погружения обрезанного конца поджелудочной железы в просвет тонкой кишки (рис. 16).
Рисунок 15. Интраоперационная картина: почти тотальное удаление головки поджелудочной железы (обнажен холедох, удалена ткань железы между холедохом и внутренней частью подковы двенадцатиперстной кишки)
Рисунок 16. Интраоперационная картина: панкреатоеюноанастомоз с оставшимся дистальным участком поджелудочной железы
Послеоперационное ведение
При переводе в отделение реанимации продолжается титрование растворов глюкозы. Контроль гликемии каждый час, затем каждые 2–3 ч. Целевой уровень гликемии – 4–8 ммоль/л. Экстубация проводится по состоянию ребенка, продленной вентиляции не предполагается. В первые сутки после операции проводится введение ингибиторов протеолиза, раствор октреотида вводится из расчета 5 мкг/кг/сутки 4–5 дней подкожно с панкреатостатической целью. Продленная перидуральная блокада проводится в течение 3–4 дней. Через сутки после операции проводится контроль уровня амилазы в крови, при ее низком уровне – отмена ингибиторов протеолиза и постепенная отмена октреотида. С началом энтерального кормления ребенок переводится в отделение детской хирургии, постепенно снижается доза углеводной нагрузки при расширении объема и усвоении питания. На 7-е сутки после операции при отсутствии хирургических осложнений ребенку снимают швы, и пациент переводится в эндокринологическое отделение.
С введением в комплекс предоперационного обследования ПЭТ/КТ с 18F-ДОФА и появлением возможности проведения экспресс-биопсии поджелудочной железы интраоперационно за 2017 – февраль 2021 г. было оперировано 39 пациентов, из них у 36 (92,3%) с полным купированием гиперинсулинизма, значительным улучшением психомоторного развития. Однако 9 из них (23%) нуждаются в заместительной инсулинотерапии минимальными дозировками – это 8 детей с диффузной формой гиперинсулинизма и 1 ребенок – с атипичной формой. Интра- и послеоперационных осложнений не отмечалось. После операции отмечаются значительное улучшение психоэмоционального состояния, быстрое прогрессирование в психомоторном развитии.
Осложнения
Любое лечение ВГИ, включая медикаментозное или хирургическое, может иметь свои побочные эффекты. Длительное использование диазоксида связано с гипертрихозом, и это может ограничивать его длительное использование (рис. 7). Октреотид может вызывать широкий спектр побочных эффектов, включая желудочнокишечные расстройства (боль в животе, тошноту, вздутие и диарею), а также подавление гормона роста, тиреотропного и адренокортикотропного гормонов. При хирургическом лечении рецидив гипогликемии может возникнуть в результате недостаточной хирургической резекции диффузной формы заболевания или из-за неудачной локализации при очаговой форме, которую не удается удалить полностью. Стойкая гипогликемия в послеоперационном периоде может потребовать повторного хирургического вмешательства. К другим хирургическим осложнениям относятся послеоперационная инфекция, кровотечение и операционная травма желчного протока. Для уменьшения частоты послеоперационных осложнений хирургическое лечение при ВГИ должно проводиться в тех единичных центрах в мире, где имеется наибольший опыт лечения таких пациентов.
Заключение
Врожденный гиперинсулинизм – настолько редкое заболевание, что только несколько детских хирургических центров в мире могут накопить достаточный опыт лечения таких пациентов. Хирургический опыт необходим для получения стабильно хороших результатов и низкого уровня осложнений. ПЭТ/КТ с 18F-ДОФА, несомненно, является наиболее точным и клинически значимым исследованием для предоперационной дифференциальной диагностики диффузных и фокальных форм ВГИ. Лапароскопические методы оперативного лечения вполне применимы при лечении пациентов с ВГИ и, вероятно, станут стандартом для первоначальной биопсии и диагностики и, возможно, для резекции очаговых поражений, за исключением головки поджелудочной железы. Вероятно, дальнейшее развитие генетики и молекулярной биологии ВГИ приведет к возможности наилучшей диагностики, классификации и лечения и в некоторых случаях исчезнет необходимость хирургического вмешательства. В настоящее время новорожденных и младенцев с гиперинсулинизмом всегда следует лечить безотлагательно и очень агрессивно с помощью внутривенной инфузии глюкозы (через центральный венозный катетер), применения доступных гликемических препаратов и частого мониторинга уровня глюкозы в крови, поскольку отдаленные последствия гипогликемии для нервной системы могут быть серьезными, разрушительными и необратимыми. В связи с этим решение о необходимости оперативного лечения должно быть максимально ранним.
Достижения в области молекулярной генетики, методы визуализации (ПЭТ/КТ), медикаментозная терапия и хирургическое лечение дали возможность лучше контролировать гипогликемию и таким образом улучшили результаты лечения этих детей. В случае успешного консервативного лечения в дальнейшем у некоторых пациентов возможна его постепенная отмена (к 4–16 годам и позже). При эффективности консервативной терапии после длительного медикаментозного лечения с постоянным дробным кормлением и контролем уровня сахара в крови с возрастом у части неоперированных пациентов (до 23%) с ВГИ описано развитие сахарного диабета [62, 66]. Среди детей, перенесших субтотальную панкреатэктомию, около 27–36% страдают от инсулинзависимого сахарного диабета как сразу после операции, так и с возможным развитием в течение 8–40 лет после операции; в противовес этому другие 2–41% продолжают нуждаться в лечении гипогликемии [62, 63, 67, 68]. Несмотря на дифференциальную диагностику различных форм ВГИ методом ПЭТ/КТ с 18F-ДОФА перед операцией [69], окончательно подтвердить тот или иной морфологический вариант возможно только при гистологическом исследовании. Необходима срочная интраоперационная гистологическая верификация поражения поджелудочной железы, поскольку именно на основании гистологии хирург определяет объем хирургического вмешательства, тем более визуально и пальпаторно определить точную локализацию пораженного участка часто не представляется возможным. Задержка психомоторного развития отмечается у 30–60% всех пациентов с ВГИ, а у 15–25% наблюдается тяжелое органическое поражение головного мозга, включая эпилепсию [46, 49, 50, 67, 68]. Своевременная диагностика и адекватное лечение позволяют предотвратить развитие тяжелых неврологических осложнений [69]. При неэффективности консервативной терапии необходимо хирургическое лечение. Частичная панкреатэктомия при фокальных, субтотальная (95%) при атипичных и почти тотальная (98–99%) при диффузных формах ВГИ позволяют справиться с гипогликемиями и предотвратить поражение центральной нервной системы новорожденных и младенцев.
Список литературы / References
McQuarrie I. Idiopathic Spontaneously Occurring Hypoglycemia in Infants; Clinical Significance of Problem and Treatment. Am J Dis Child. 1954;87(4):399-428. https://doi.org/10.1001/archpedi.1954.02050090387001.- Menni F., de Lonlay P., Sevin C., Touati G., Peigné C., Barbier V. et al. Neurologic Outcomes of 90 Neonates and Infants with Persistent Hyperinsulinemic Hypoglycemia. Pediatrics. 2001;107(3):476-479. https://doi.org/10.1542/peds.107.3.476.
- Helleskov A., Melikyan M., Globa E., Shcherderkina I., Poertner F., Larsen A.M. et al. Both Low Blood Glucose and Insufficient Treatment Confer Risk of Neurodevelopmental Impairment in Congenital Hyperinsulinism: A Multinational Cohort Study. Front Endocrinol (Lausanne). 2017;8:156. https://doi.org/10.3389/fendo.2017.00156.
- Hussain K., Blankenstein O., De Lonlay P., Christesen H. Hyperinsulinaemic hypoglycaemia: Biochemical basis and the importance of maintaining normoglycaemia during management. Arch Dis Child. 2007;92(7):568-570. https://doi.org/10.1136/adc.2006.115543.
- Mathew P.M.., Young J.M., AbuOsba Y.K., Mulhern B.D., Hammoudi S., Hamdan J.A., Sa’di A.R. Persistent Neonatal Hyperinsulinism. Clin Pediatr (Phila). 1988;27(3):148-151. https://doi.org/10.1177/000992288802700307.
- Kapoor R.R., Flanagan S.E., James C., Shield J., Ellard S., Hussain K. Hyperinsulinaemic Hypoglycaemia. Arch Dis Child. 2009;94:450-457. https://doi.org/10.1136/adc.2008.148171.
- Prentki M., Matschinsky F.M., Madiraju S.R.M. Metabolic Signaling in FuelInduced Insulin Secretion. Cell Metab. 2013;18(2):162-185. https://doi.org/10.1016/j.cmet.2013.05.018.
- Brun T., Roche E., AssimacopoulosJeannet F., Corkey B.E., Kim K.-H., Prentki M. Evidence for an Anaplerotic/MalonylCoA Pathway in Pancreatic βCell Nutrient Signaling. Diabetes. 1996;45(2):190-198. https://doi.org/10.2337/diab.45.2.190.
- Deeney J.T., Gromada J., Hoy M., Olsen H.L., Rhodes C.J., Prentki M. et al. Acute Stimulation with Long Chain AcylCoA Enhances Exocytosis in InsulinSecreting Cells (HIT T-15 and NMRI BetaCells). J Biol Chem. 2000;275(13):9363-9368. https://doi.org/10.1074/jbc.275.13.9363.
- Petersen O.H., Findlay I. Electrophysiology of the Pancreas. Physiol Rev. 1987;67(3):1054-1116. https://doi.org/10.1152/physrev.1987.67.3.1054.
- Shimono D., Fujimoto S., Mukai E., Takehiro M., Nabe K., Radu R.G. et al. ATP Enhances Exocytosis of Insulin Secretory Granules in Pancreatic Islets under Ca2+ - Depleted Condition: Diabetes Res Clin Pract. 2005;69(3):216- 223. https://doi.org/10.1016/j.diabres.2005.01.010.
- SaintMartin C., Arnoux J.-B., de Lonlay P., BellannéChantelot C. KATP Channel Mutations in Congenital Hyperinsulinism. Semin Pediat Surg. 2011;20(1):18-22. https://doi.org/10.1053/j.sempedsurg.2010.10.012.
- Thornton P.S., MacMullen C., Ganguly A., Ruchelli E., Steinkrauss L., Crane A. et al. Clinical and Molecular Characterization of a Dominant Form of Congenital Hyperinsulinism Caused by a Mutation in the HighAffinity Sulfonylurea Receptor. Diabetes. 2003;52(9):2403-2410. https://doi.org/10.2337/diabetes.52.9.2403.
- Huopio H., Reimann F., Ashfield R., Komulainen J., Lenko H.-L., Rahier J. et al. Dominantly Inherited Hyperinsulinism Caused by a Mutation in the Sulfonylurea Receptor Type 1. J Clin Invest. 2000;106(7):897-906. https://doi.org/10.1172/JCI9804.
- Nessa A., Hussain K., Thomas A.M., Tinker A., Aziz Q.H., Harmer S.C. Molecular Mechanisms of Congenital Hyperinsulinism Due to Autosomal Dominant Mutations in ABCC8. Hum Mol Genet. 2015; 24(18):5142-5153. https://doi.org/10.1093/hmg/ddv233.
- Flanagan S., Kapoor R., Banerjee I., Hall C., Smith V., Hussain K., Ellard S. Dominantly Acting ABCC8 Mutations in Patients with Medically Unresponsive Hyperinsulinaemic Hypoglycaemia. Clin Genet. 2011;79(6):582-587. https://doi.org/10.1111/j.1399-0004.2010.01476.x.
- Sempoux C., Guiot Y., Rahier J., Fekete C., Aigrain Y., Capito C. et al. Morphological Mosaicism of the Pancreatic Islets: A Novel Anatomopathological Form of Persistent Hyperinsulinemic Hypoglycemia of Infancy. J Clin Endocrinol Metab. 2011;96(12):3785-3793. https://doi.org/10.1210/jc.2010-3032.
- De Lonlay P., Fournet J.C., Rahier J., GrossMorand M.S., PoggiTravert F., Foussier V. et al. Somatic Deletion of the Imprinted 11p15 Region in Sporadic Persistent Hyperinsulinemic Hypoglycemia of Infancy Is Specific of Focal Adenomatous Hyperplasia and Endorses Partial Pancreatectomy. J Clin Invest. 1997;100(4):802-807. https://doi.org/10.1172/JCI119594.
- Damaj L., le Lorch M., Verkarre V., Werl C., Hubert L., NihoulFékété C et al. Chromosome 11p15 Paternal Isodisomy in Focal Forms of Neonatal Hyperinsulinism. J Clin Endocrinol Metab. 2008;93(12):4941-4947. https://doi.org/10.1210/jc.2008-0673.
- Stanley C.A., Lieu Y.K., Hsu B.Y., Burlina A.B., Greenberg C.R., Hopwood N.J. et al. Hyperinsulinism and Hyperammonemia in Infants with Regulatory Mutations of the Glutamate Dehydrogenase Gene. N Engl J Med. 1998;338(19):1352-1357. https://doi.org/10.1056/NEJM199805073381904.
- Weinzimer S.A., Stanley C.A., Berry G.T., Yudkoff M., Tuchman M., Thornton P.S. A Syndrome of Congenital Hyperinsulinism and Hyperammonemia. J Pediatr. 1997;130(4):661-664. https://doi.org/10.1016/s0022-3476(97)70256-7.
- Fang J., Hsu B.Y., MacMullen C.M., Poncz M., Smith T.J., Stanley C.A. Expression, Purification and Characterization of Human Glutamate Dehydrogenase (GDH) Allosteric Regulatory Mutations. Biochem J. 2002;363(1):81-87. https://doi.org/10.1042/0264-6021:3630081.
- Kapoor R.R., James C., Flanagan S.E., Ellard S., Eaton S., Hussain K. 3-HydroxyacylCoenzyme A Dehydrogenase Deficiency and Hyperinsulinemic Hypoglycemia: Characterization of a Novel Mutation and Severe Dietary Protein Sensitivity. J Clin Endocrinol Metab. 2009;94(7):2221-2225. https://doi.org/10.1210/jc.2009-0423.
- Clayton P.T., Eaton S., AynsleyGreen A., Edginton M., Hussain K., Krywawych S. et al. Hyperinsulinism in ShortChain L-3-HydroxyacylCoA Dehydrogenase Deficiency Reveals the Importance of BetaOxidation in Insulin Secretion. J Clin Invest. 2001;108(3):457-465. https://doi.org/10.1172/JCI11294.
- Martins E., Cardoso M.L., Rodrigues E., Barbot C., Ramos A., Bennett M.J. et al. ShortChain 3-HydroxyacylCoA Dehydrogenase Deficiency: The Clinical Relevance of an Early Diagnosis and Report of Four New Cases. J Inherit Metab Dis. 2011;34(3):835-842. https://doi.org/10.1007/s10545-011-9287-7.
- Li C., Chen P., Palladino A., Narayan S., Russell L.K., Sayed S. et al. Mechanism of Hyperinsulinism in ShortChain 3-HydroxyacylCoA Dehydrogenase Deficiency Involves Activation of Glutamate Dehydrogenase. J Biol Chem. 2010;285(41):31806-31818. https://doi.org/10.1074/jbc.M110.123638.
- Thornton P.S., Stanley C.A., De Leon D.D., Harris D., Haymond M.W., Hussain K. et al. Pediatric Endocrine Society. Recommendations from the Pediatric Endocrine Society for Evaluation and Management of Persistent Hypoglycemia in Neonates, Infants, and Children. J Pediatr. 2015;167(2):238-245. https://doi.org/10.1016/j.jpeds.2015.03.057.
- Sempoux C., Guiot Y., Jaubert F., Rahier J. Focal and Diffuse Forms of Congenital Hyperinsulinism: The Keys for Differential Diagnosis. Endocr Pathol. 2004;15(3):241-246. https://doi.org/10.1385/ep:15:3:241.
- Han B., Mohamed Z., Estebanez M.S., Craigie R.J., Newbould M., Cheesman E. et al. Atypical Forms of Congenital Hyperinsulinism in Infancy Are Associated With Mosaic Patterns of Immature Islet Cells. J Clin Endocrinol Metab. 2017;102(9):3261-3267. https://doi.org/10.1210/jc.2017-00158.
- Ryan F., Devaney D., Joyce C., Nestorowicz A., Permutt M.A., Glaser B. et al. Hyperinsulinism: Molecular Aetiology of Focal Disease. Arch Dis Child. 1998;79(5):445-447. https://doi.org/10.1136/adc.79.5.445.
- Houghton J., Banerjee I., Shaikh G., Jabbar S., Laver T.W., Cheesman E. et al. Unravelling the Genetic Causes of Mosaic Islet Morphology in Congenital Hyperinsulinism. J Pathol Clin Res. 2020;6(1):12-16. https://doi.org/ 10.1002/cjp2.144.
- Сухоцкая А.А., Баиров В.Г., Перминова А.А., Митрофанова Л.Б., Никитина И.Л., Рыжкова Д.В., Амидхонова С.А. Зависимость объема резекции поджелудочной железы при врожденном гиперинсулинизме от данных ПЭТ-томографии и экспрессбиопсии. Детская хирургия. 2021;25(1):11-18. https://doi.org/10.18821/1560-9510-2021-25-1-11-18.
- Fékété C.N., de Lonlay P., Jaubert F., Rahier J., Brunelle F., Saudubray J.M. The Surgical Management of Congenital Hyperinsulinemic Hypoglycemia in Infancy. J Pediatr Surg. 2004;39(3):267-269. https://doi.org/10.1016/j.jpedsurg.2003.11.004.
- Adzick N.S., Thornton P.S., Stanley C.A., Kaye R.D., Ruchelli E. A Multidisciplinary Approach to the Focal Form of Congenital Hyperinsulinism Leads to Successful Treatment by Partial Pancreatectomy. J Pediatr Surg. 2004;39(3): 270-275. https://doi.org/10.1016/j.jpedsurg.2003.11.019.
- Blomberg B.A., Moghbel M.C., Saboury B., Stanley C.A., Alavi A. The Value of Radiologic Interventions and (18) F-DOPA PET in Diagnosing and Localizing Focal Congenital Hyperinsulinism: Systematic Review and MetaAnalysis. Mol Imaging Biol. 2013;15(1):97-105. https://doi.org/10.1007/s11307-012-0572-0.
- Dubois J., Brunelle F., Touati G., Sebag G., Nuttin C., Thach T. et al. Hyperinsulinism in Children: Diagnostic Value of Pancreatic Venous Sampling Correlated with Clinical, Pathological and Surgical Outcome in 25 Cases. Pediatr Radiol. 1995;25(7):512-516. https://doi.org/10.1007/BF02015782.
- Ferry R.J. Jr, Kelly A., Grimberg A., KooMcCoy S., Shapiro M.J., Fellows K.E. et al. CalciumStimulated Insulin Secretion in Diffuse and Focal Forms of Congenital Hyperinsulinism. J Pediatr. 2000;137(2):239-246. https://doi.org/10.1067/mpd.2000.107386.
- Ribeiro M.J., Boddaert N., BellannéChantelot C., Bourgeois S., Valayannopoulos V., Delzescaux T. et al. The Added Value of [18F] fluoroLDOPA PET in the Diagnosis of Hyperinsulinism of Infancy: A Retrospective Study Involving 49 Children. Eur J Nucl Med Mol Imaging. 2007;34(12):2120-2128. https://doi.org/10.1007/s00259-007-0498-y.
- De Lonlay P., SimonCarre A., Ribeiro M.J., Boddaert N., Giurgea I., Laborde K. et al. Congenital Hyperinsulinism: Pancreatic [18F]FluoroL-Dihydroxyphenylalanine (DOPA) Positron Emission Tomography and Immunohistochemistry Study of DOPA Decarboxylase and Insulin Secretion. J Clin Endocrinol Metab. 2006;91(1):933-940. https://doi.org/10.1210/jc.2005-1713.
- Koopmans K.P., Neels O.N., Kema I.P., Elsinga P.H., Links T.P., de Vries E.G. et al. Molecular Imaging in Neuroendocrine Tumors: Molecular Uptake Mechanisms and Clinical Results. Crit Rev Oncol Hematol. 2009;71(3): 199-213. https://10.1016/j.critrevonc.2009.02.009.
- Lindstrom P. AromaticL-AminoAcid Decarboxylase Activity in Mouse Pancreatic Islets. Biochim Biophys Acta. 1986;884(2):276-281. https://doi.org/10.1016/0304-4165(86)90174-1.
- Ericson L.E., Hakanson R., Lundquist I. Accumulation of Dopamine in Mouse Pancreatic B-Cells Following Injection of L-DOPA. Localization to Secretory Granules and Inhibition of Insulin Secretion. Diabetologia. 1977;13(2):117-124. https://doi.org/10.1007/BF00745138.
- Farino Z.J., Morgenstern T.J., Maffei A., Quick M., De Solis A.J., Wiriyasermkul P. et al. New Roles for Dopamine D2 and D3 Receptors in Pancreatic Beta Cell Insulin Secretion. Mol Psychiatry. 2020;25(9):2070-2085. https://doi.org/10.1038/s41380-018-0344-6.
- Rubi B., Ljubicic S., Pournourmohammadi S., Carobbio S., Armanet M., Bartley C. et al. Dopamine D2-Like Receptors Are Expressed in Pancreatic Beta Cells and Mediate Inhibition of Insulin Secretion. J Biol Chem. 2005;280(44):36824-36832. https://doi.org/10.1074/jbc.M505560200.
- Ustione A., Piston D.W. Dopamine Synthesis and D3 Receptor Activation in Pancreatic βCells Regulates Insulin Secretion and Intracellular [Ca(2+)] Oscillations. Mol Endocrinol. 2012;26(11):1928-1940. https://doi.org/10.1210/me.2012-1226.
- Santhanam P., Taïeb D. Role of 18F-FDOPA PET/CT Imaging in Endocrinology. Clin Endocrinol (Oxf). 2014;81(6):789-798. https://doi.org/10.1111/cen.12566.
- Yang J., Hao R., Zhu X. Diagnostic Role of 18F-Dihydroxyphenylalanine Positron Emission Tomography in Patients with Congenital Hyperinsulinism: A MetaAnalysis. Nucl Med Commun. 2013;34(4):347-353. https://doi.org/10.21569/2222-7415-2017-7-3-144-152.
- Meintjes M., Endozo R., Dickson J., Erlandsson K., Hussain K., Townsend C. et al. 18F-DOPA PET and Enhanced CT Imaging for Congenital Hyperinsulinism: Initial UK Experience from a Technologist’s Perspective. Nucl Med Commun. 2013;34(6):601-608. https://doi.org/10.1097/MNM.0b013e32836069d0.
- Chondrogiannis S., Grassetto G., Marzola M.C., Rampin L., Massaro A., Bellan E. et al. 18F-DOPA PET/CT Biodistribution Consideration in 107 Consecutive Patients with Neuroendocrine Tumours. Nucl Med Commun. 2012;33(2): 179-184. https://doi.org/10.1097/MNM.0b013e32834e0974.
- Mohnike K., Blankenstein O., Christesen H.T., De Lonlay J., Hussain K., Koopmans K.P. et al. Proposal for a Standardized Protocol for 18F-DOPAPET (PET/CT) in Congenital Hyperinsulinism. Horm Res. 2006;66(1):40-42. https://doi.org/10.1159/000093471.
- Hardy O.T., HernandezPampaloni M., Saffer J.R., Scheuermann J.S., Ernst L.M., Freifelder R. et al. Accuracy of [18F]Fluorodopa Positron Emission Tomography for Diagnosing and Localizing Focal Congenital Hyperinsulinism. J Clin Endocrinol Metab. 2007;92(12):4706-47011. https://doi.org/10.1210/jc.2007-1637.
- Otonkoski T., Näntö-Salonen K., Seppänen M., Veijola R., Huopio H., Hussain K. et al. Noninvasive Diagnosis of Focal Hyperinsulinism of Infancy with [18F]-DOPA Positron Emission Tomography. Diabetes. 2006;55(1):13-18. https://doi.org/10.2337/diabetes.55.01.06.db05-1128.
- Treglia G., Mirk P., Giordano A., Rufini V. Diagnostic Performance of Fluorine-18-Dihydroxyphenylalanine Positron Emission Tomography in Diagnosing and Localizing the Focal Form of Congenital Hyperinsulinism: A Metaanalysis. Pediatr Radiol. 2012;42(11):1372-1379. https://doi.org/10.1007/s00247-012-2459-2.
- Barthlen W., Blankenstein O., Mau H., Koch M., Hohne C., Mohnike W. et al. Evaluation of [18F]FluoroL-DOPA Positron Emission TomographyComputed Tomography for Surgery in Focal Congenital Hyperinsulinism. J Clin Endocrinol Metab. 2008;93(3):869-875. https://doi.org/10.1210/jc.2007-2036.
- Yang J., Yuan L., Meeks J.K., Zhang N., Li C., Hao R. 18F-DOPA Positron Emission Tomography/Computed Tomography Application in Congenital Hyperinsulinism. J Pediatr Endocrinol Metab. 2012;25(7-8):619-622. https://doi.org/10.1515/jpem-2012-0114.
- Jodal A., Schibli R., Béhé M. Targets and Probes for NonInvasive Imaging of βCells. Eur J Nucl Med Mol Imaging. 2017;44(4):712-727. https://doi.org/10.1007/s00259-016-3592-1.
- Laje P., Stanley C.A., Palladino A.A., Becker S.A., Adzick N.S. Pancreatic Head Resection and Roux-enY Pancreaticojejunostomy for the Treatment of the Focal Form of Congenital Hyperinsulinism. J Pediatr Surg. 2012;47(1):130-135. https://doi.org/10.1016/j.jpedsurg.2011.10.032.
- Lovvorn H.N., Nance M.L., Ferry R.J. Jr., Stolte L., Baker L., O’Neill J.A. Jr et al. Congenital Hyperinsulinism and the Surgeon: Lessons Learned Over 35 Years. J Pediatr Surg. 1999;34(5):786-792. https://doi.org/10.1016/s0022-3468(99)90374-3.
- Губаева Д.Н., Меликян М.А., Рыжкова Д.В., Пойда М.Д., Баиров В.Г., Сухоцкая А.А. и др. Клинические, генетические и радионуклидные характеристики пациентов с фокальной формой врожденного гиперинсулинизма. Проблемы эндокринологии. 2019;65(5):319-329. https://doi.org/10.14341/probl10317.
- Lord K., De León D.D. Hyperinsulinism in the Neonate. Clin Perinatol. 2018;45(1):61-74. https://doi.org/10.1016/j.clp.2017.10.007.
- MazorAronovitch K., Landau H., Gillis D. Surgical versus NonSurgical Treatment of Congenital Hyperinsulinism. Pediatr Endocrinol Rev. 2009;6(3):424-430. Available at: https://pubmed.ncbi.nlm.nih.gov/19396028/.
- Ismail D., Werther G. Persistent Hyperinsulinaemic Hypoglycaemia of Infancy: 15 Years’ Experience at the Royal Children’s Hospital (RCH), Melbourne. J Pediatr Endocrinol Metab. 2005;18(11):1103-1109. https://doi.org/10.1515/jpem.2005.18.11.1103.
- Meissner T., Wendel U., Burgard P., Schaetzle S., Mayatepek E. LongTerm FollowUp of 114 Patients with Congenital Hyperinsulinism. Eur J Endocrinol. 2003;149(1):43-51. https://doi.org/10.1530/eje.0.1490043.
- Graham E.A., Hartmann A.F. Subtotal Resection of the Pancreas for Hypoglycaemia. Surg Gynecol Obstet. 1934;59:474-479.
- Adzick N.S., De León D.D., States L.J., Lord K., Bhatti T.R., Becker S.A., Stanley C.A. Surgical Treatment of Congenital Hyperinsulinism: Results from 500 Pancreatectomies in Neonates and Children. J Pediatr Surg. 2019;54(1):27-32. https://doi.org/10.1016/j.jpedsurg.2018.10.030.
- Gussinyer M., Clemente M., Cebrián R., Yeste D., Albisu M., Carrascosa A. Glucose Intolerance and Diabetes Are Observed in the LongTerm FollowUp of Nonpancreatectomized Patients with Persistent Hyperinsulinemic Hypoglycemia of Infancy due to Mutations in the ABCC8 Gene. Diabetes Care. 2008;31(6):1257-1259. https://doi.org/10.2337/dc07-2059.
- Lord K., Radcliffe J., Gallagher P.R., Adzick N.S., Stanley C.A., De León D.D. High Risk of Diabetes and Neurobehavioral Deficits in Individuals with Surgically Treated Hyperinsulinism. J Clin Endocrinol Metab. 2015;100(11):4133-4139. https://doi.org/10.1210/jc.2015-2539.
- Lord K., Dzata E., Snider K.E., Gallagher P.R., De León D.D. Clinical Presentation and Management of Children With Diffuse and Focal Hyperinsulinism: A Review of 223 Cases. J Clin Endocrinol Metab. 2013;98(11):E1786-E1789. https://doi.org/10.1210/jc.2013-2094.
- Ni J., Ge J., Zhang M., Hussain K., Guan Y., Cheng R. et al. Genotype and Phenotype Analysis of a Cohort of Patients with Congenital Hyperinsulinism Based on DOPA-PET CT Scanning. Eur J Pediatr. 2019;178(8):1161-1169. https://doi.org/10.1007/s00431-019-03408-6.
Другие новости
Что будет, если переедать семечками подсолнечника ежедневно

Увлекаясь поеданием семечек подсолнечника ежедневно, многие не знают, что 300 граммов этого продукта равносильны...
Sanofi намерена создать одного из крупнейших производителей

В статье обсуждаются вопросы, касающиеся лечения больных c персистирующей формой фибрилляц......
Новогодний стол для язвенников: что включить в меню, чтобы

Опытные хозяйки уже начинают планировать меню к Новому году. К сожалению, многие традиционные блюда не подойдут для...